La ataxia espinocerebelosa no es una sola enfermedad, sino un grupo de trastornos genéticos que alteran la coordinación, el equilibrio y, con frecuencia, el habla o la deglución. Cuando aparece en una familia, lo importante no es solo ponerle nombre al cuadro, sino entender qué patrón de herencia puede haber detrás, qué pruebas lo confirman y qué apoyos cambian de verdad la evolución cotidiana.
En este artículo repaso qué son estas ataxias, cuáles son sus señales más útiles para sospecharlas, cómo se estudian en genética clínica y qué opciones reales existen para el manejo, el consejo familiar y la planificación reproductiva.
Lo esencial para orientarse sin perder tiempo
- Se trata de un grupo heterogéneo de enfermedades neurogenéticas, no de un diagnóstico único.
- Lo más típico es la marcha inestable, pero también pueden aparecer disartria, nistagmo, disfagia, rigidez o temblor.
- La herencia suele ser autosómica dominante, aunque existen formas recesivas y otras variantes menos frecuentes.
- La confirmación depende sobre todo del estudio genético adecuado; una resonancia por sí sola no basta.
- El tratamiento suele ser de apoyo: rehabilitación, logopedia, terapia ocupacional y control de síntomas.
- El consejo genético importa tanto como el diagnóstico, porque cambia decisiones médicas y familiares.
Qué es este grupo de ataxias y por qué no todas evolucionan igual
Yo suelo explicarlo así: el problema no está solo en “caminar mal”, sino en una red cerebral que empieza a fallar, sobre todo en el cerebelo y en sus conexiones. Esa pérdida de precisión convierte movimientos que deberían ser automáticos en gestos torpes, inestables o descoordinados. Con el tiempo, según el subtipo, pueden añadirse problemas del habla, los ojos, la deglución o la motricidad fina.
Lo que complica este grupo es su heterogeneidad. Algunas formas debutan en la juventud, otras lo hacen en la edad adulta; unas progresan despacio, otras dan un salto funcional más rápido; y en ciertas familias la enfermedad parece “más temprana” en cada generación por un fenómeno llamado anticipación, típico de varias expansiones de repetidos en el ADN. También conviene no mezclarlo todo: no toda ataxia hereditaria pertenece a la misma familia molecular, y eso cambia bastante el enfoque diagnóstico.
Con esta base en mente, lo siguiente es reconocer las señales que suelen aparecer primero y que, en la práctica, suelen hacer saltar la sospecha clínica.
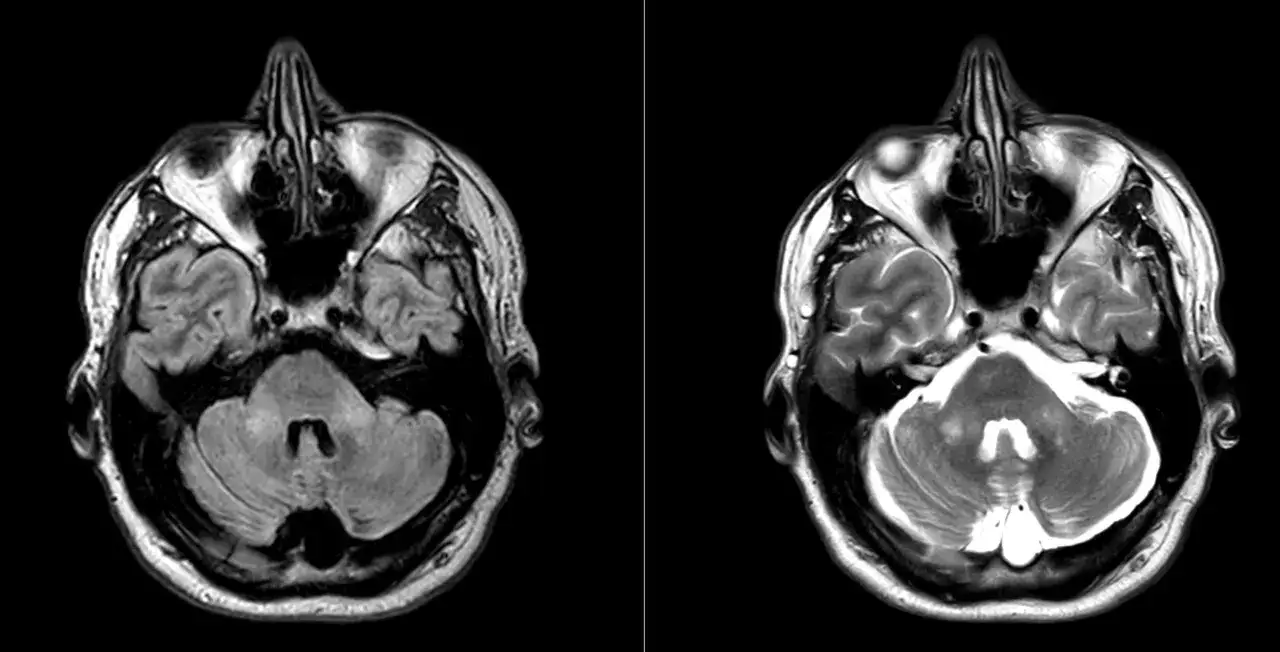
Señales que suelen aparecer primero
En consulta, la pista inicial suele ser muy prosaica: alguien empieza a tropezar más, a desviarse al girar, a escribir peor o a notar que el habla se vuelve más lenta y “pastosa”. Esa mezcla de síntomas no siempre llega junta, y a veces el entorno tarda en darle importancia porque se confunde con cansancio, edad o torpeza aislada.
Cuando quiero ordenar el cuadro, me fijo en si la alteración es principalmente de la marcha o si ya afecta a varias funciones cerebelosas a la vez. Ese matiz ayuda a decidir la rapidez del estudio y a orientar qué subtipo puede haber detrás.
| Señal | Cómo suele notarse | Por qué importa |
|---|---|---|
| Ataxia de la marcha | Pasos amplios, inseguridad al girar, caídas o sensación de ir “ebrio” sin haber bebido | Suele ser la primera manifestación y la más fácil de pasar por alto |
| Disartria | Habla entrecortada, poco precisa o con ritmo extraño | Refleja afectación de la coordinación del lenguaje motor |
| Nistagmo o diplopía | Ojos que “saltan” o visión doble | Apunta a afectación de vías oculomotoras, frecuente en varios subtipos |
| Disfagia | Tos al comer, atragantamientos, pérdida de peso o comidas muy lentas | Es relevante porque aumenta el riesgo de aspiración y desnutrición |
| Rigidez, temblor o espasticidad | Dificultad para soltar movimientos finos o sensación de piernas rígidas | Hace pensar en formas más amplias, no solo cerebelosas |
Una cosa útil: si el cuadro combina varios de estos signos, la sospecha genética sube bastante. Y cuando eso pasa, el siguiente paso no es “esperar a ver qué ocurre”, sino ordenar bien la herencia posible, porque ahí se decide gran parte del mapa familiar.
Cómo se hereda y qué significa para la familia
La gran mayoría de las formas clásicas siguen un patrón autosómico dominante: basta una copia alterada del gen para que la enfermedad pueda aparecer. En ese escenario, si uno de los progenitores está afectado, cada hijo tiene un riesgo del 50 % en cada embarazo, aunque la expresión clínica no siempre sea idéntica entre familiares. Esa variabilidad es importante, porque dos personas con la misma mutación pueden debutar a edades distintas o progresar a ritmos muy diferentes.
También existen formas autosómicas recesivas, menos frecuentes en este grupo, en las que ambos progenitores suelen ser portadores sanos. En ese caso, el riesgo de un hijo afectado es del 25 % si los dos transmiten la variante. Y además hay subtipos ligados a expansiones de tripletes, como las repeticiones CAG, donde el número de repeticiones puede influir en la edad de inicio: en términos simples, más repeticiones suelen asociarse con un comienzo más precoz y, a menudo, con una evolución más intensa.
| Patrón | Qué suele implicar | Riesgo para la descendencia | Pista práctica |
|---|---|---|---|
| Autosómico dominante | Una sola copia alterada puede bastar para producir la enfermedad | 50 % por embarazo si uno de los progenitores está afectado | Suele haber más de una generación implicada |
| Autosómico recesivo | Se necesitan dos copias alteradas | 25 % si ambos progenitores son portadores | Pueden aparecer hermanos afectados con padres sanos |
| Expansión de repetidos | El ADN repite un fragmento más veces de lo normal | Variable según el gen y la longitud de la repetición | Puede existir anticipación en generaciones sucesivas |
Yo suelo insistir en un detalle que cambia mucho la estrategia: que no haya antecedentes claros no descarta una enfermedad hereditaria. A veces el árbol familiar está “vacío” porque nadie fue diagnosticado, porque el inicio fue leve o porque hubo una mutación nueva. Con eso claro, el estudio deja de ser una búsqueda genérica y pasa a ser una investigación bien dirigida.
Cómo se confirma el diagnóstico sin perder tiempo
En España, el circuito más útil suele pasar por neurología y genética clínica. No porque haya que acumular especialistas, sino porque el diagnóstico correcto depende de unir la exploración, la historia familiar y el tipo exacto de prueba molecular. Aquí es donde mucha gente pierde tiempo: se hace una prueba demasiado general, el resultado sale incompleto y luego se concluye, con demasiada prisa, que “no hay nada genético”.
Yo no daría por cerrado un caso con un primer panel negativo si la clínica sigue encajando. En estas enfermedades, el método importa tanto como el gen, porque no todos los análisis detectan bien las expansiones de repetidos ni todos los paneles cubren los mismos subtipos.
- Historia clínica y familiar detallada: edad de inicio, caídas, problemas del habla, visión, deglución y antecedentes en otros parientes.
- Exploración neurológica: busca signos cerebelosos, oculomotores, piramidales o sensitivos que orienten el subtipo.
- Resonancia magnética: puede mostrar atrofia cerebelosa u otros cambios, pero no confirma por sí sola el diagnóstico.
- Estudio genético dirigido: paneles de ataxias, pruebas de expansiones CAG o, si hace falta, exoma o genoma.
- Consejo genético: antes y después del resultado, para explicar qué significa, qué no significa y a quién más afecta.
Un matiz técnico importante: cuando sospecho una expansión de repetidos, pido expresamente que el laboratorio confirme que el método la detecta bien. Ese detalle evita falsos negativos “por diseño” y ahorra vueltas innecesarias. Una vez que el nombre está bien puesto, la pregunta pasa a ser cómo reducir complicaciones y mantener función el mayor tiempo posible.
Qué tratamiento y apoyo tienen más impacto
La realidad, sin adornos, es que la mayoría de estas ataxias no tienen una cura consolidada. Eso no significa que no haya nada que hacer. Al contrario: el manejo correcto puede cambiar mucho la autonomía, la seguridad en casa y la calidad de vida. Lo que mejor funciona suele ser un enfoque multidisciplinar y bastante pragmático, sin esperar milagros farmacológicos que hoy todavía no existen para la mayoría de subtipos.
Yo separo el tratamiento en dos planos: el control de síntomas y la prevención de complicaciones. Esa división ayuda a no obsesionarse solo con la marcha, cuando en realidad el habla, la deglución o las caídas suelen ser lo que más deteriora el día a día.
- Fisioterapia para equilibrio, marcha, fuerza y estrategias de compensación.
- Logopedia para disartria y, sobre todo, para valorar y entrenar la deglución.
- Terapia ocupacional para adaptar actividades, utensilios, ropa, baño y cocina.
- Ayudas técnicas como bastón, andador o silla, cuando caminar deja de ser seguro.
- Control de síntomas concretos si hay espasticidad, temblor, dolor, ansiedad, insomnio o problemas oculares.
- Vigilancia nutricional si aparece disfagia, pérdida de peso o tos con las comidas.
La parte que más se subestima, y que yo casi siempre remarco, es la rehabilitación temprana. No se trata de “arreglar” la enfermedad, sino de ganar tiempo funcional y reducir lesiones evitables. Y cuando hay antecedentes en casa, además del tratamiento aparecen decisiones familiares y éticas que conviene encarar con calma.
Decisiones familiares, pruebas genéticas y bioética
Este es el punto donde la medicina personalizada se cruza con la bioética. Saber la variante causal puede ser útil para el seguimiento, para la familia y para la planificación reproductiva, pero no todo el mundo quiere conocer la misma información ni en el mismo momento. Yo separo aquí lo médico de lo personal, porque mezclarlo suele generar decisiones pobres o, peor todavía, decisiones apresuradas.
El asesoramiento genético sirve para explicar opciones reales, no para empujar a una respuesta concreta. En una familia con una ataxia hereditaria, puede ayudar a decidir si tiene sentido un estudio predictivo en un adulto asintomático, si conviene revisar a hermanos o si se plantea una estrategia reproductiva distinta.
- Prueba predictiva en adultos: útil cuando la persona quiere conocer su riesgo y entiende bien sus implicaciones.
- Menores de edad: la decisión se individualiza; si no hay una intervención preventiva clara en la infancia, suele tratarse con mucha prudencia.
- Diagnóstico prenatal: opción posible en algunas familias si ya se conoce la variante causal.
- Diagnóstico genético preimplantacional: puede plantearse en determinados casos para evitar la transmisión.
- Privacidad y apoyo psicológico: conocer el resultado no siempre es sencillo, y el acompañamiento importa.
En la práctica, lo que más agradecen muchas familias no es solo el informe del laboratorio, sino una explicación serena de qué se puede hacer con ese resultado y qué no. Con eso encima de la mesa, lo más útil es mirar hacia adelante y actuar pronto donde realmente cambia el pronóstico funcional.
Lo que cambia de verdad el día a día
Si hay una idea que me gustaría dejar clara, es esta: en este grupo de enfermedades, llegar antes al diagnóstico y empezar antes la rehabilitación suele pesar más que el nombre exacto del subtipo. El subtipo importa, sí, pero en la vida real marcan más la disfagia, las caídas, la adaptación del hogar y la red de seguimiento que el número que aparece en la historia clínica.
- Consulta antes de que las caídas sean frecuentes: una valoración temprana permite ajustar rehabilitación y ayudas.
- Pregunta por la técnica de análisis genético: no todos los estudios detectan lo mismo.
- Vigila la deglución: tos al comer, atragantamientos o pérdida de peso no son detalles menores.
- Adapta la casa pronto: buena iluminación, retirada de obstáculos y apoyos en baño y pasillos reducen riesgos.
- Guarda un árbol familiar claro: tres generaciones bien recogidas orientan más de lo que parece.
Si hay un mensaje práctico que merece quedarse, es que estas ataxias no se gestionan bien desde la espera pasiva. Cuando el estudio genético, la rehabilitación y el consejo familiar se coordinan, el paciente deja de ir a remolque del problema y empieza a tomar decisiones con más margen y menos incertidumbre.