La monosomía X, más conocida como síndrome de Turner, es una alteración cromosómica que cambia el crecimiento, la pubertad y el funcionamiento de varios órganos. No se entiende bien si se mira solo como un resultado de laboratorio: el valor real está en reconocer qué complicaciones puede producir, cómo se confirma y qué seguimiento marca la diferencia. En esta guía me centro en lo que suele buscar quien necesita una explicación clara: causas, señales, diagnóstico y decisiones médicas útiles.
Lo esencial que conviene tener claro desde el inicio
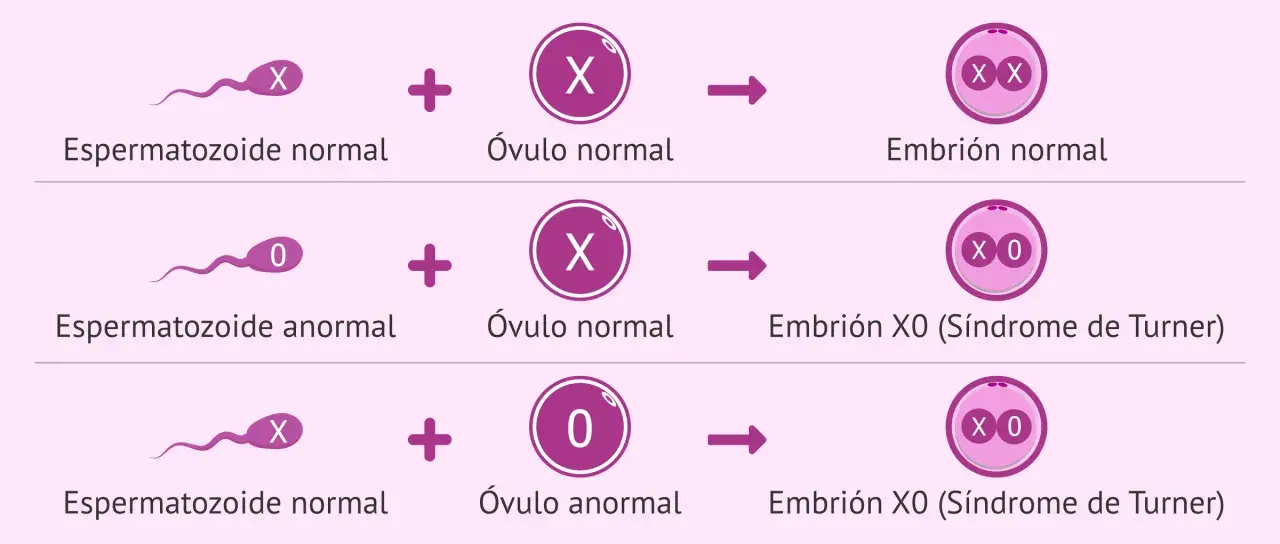
- Es una aneuploidía del cromosoma X: falta todo o parte de un X, y la forma clásica es 45,X.
- No todas las pacientes presentan el mismo cuadro; el mosaicismo y las variantes estructurales cambian mucho la expresión clínica.
- Las pistas más frecuentes son talla baja, pubertad retrasada o ausente y, en algunos casos, cardiopatías o alteraciones renales.
- El diagnóstico se confirma con cariotipo; si la sospecha sigue, hay que ampliar el estudio para no pasar por alto un mosaicismo.
- No existe una cura, pero sí un seguimiento muy eficaz con endocrinología, cardiología, genética y, según la edad, ginecología.
- Antes de valorar embarazo, la revisión cardiológica es obligatoria por el riesgo aórtico.
Qué significa la monosomía X y por qué no todas las pacientes son iguales
Lo primero es entender la base genética. En el síndrome de Turner falta todo o parte de un cromosoma X, así que se trata de una aneuploidía, es decir, una alteración en el número de cromosomas. MedlinePlus resume bien esta idea: no siempre falta el cromosoma completo, y eso explica por qué el cuadro puede ir desde signos muy visibles hasta formas mucho más sutiles.
La variante clásica es 45,X, en la que las células tienen un solo cromosoma X. Pero no es la única presentación. También existen el mosaicismo, donde unas células son 45,X y otras tienen un cariotipo habitual, y las formas estructurales, en las que el segundo X está incompleto, reorganizado o en anillo. Yo no suelo explicar el síndrome como una entidad rígida, porque en la práctica clínica no lo es: el patrón cromosómico orienta, pero el impacto real depende de qué tejidos se hayan afectado y con qué intensidad.
| Variante cromosómica | Qué ocurre | Qué suele implicar |
|---|---|---|
| 45,X clásico | Falta un cromosoma X en la mayoría o en todas las células estudiadas. | Mayor probabilidad de talla baja, pubertad ausente o incompleta e insuficiencia ovárica. |
| Mosaicismo 45,X/46,XX | Conviven células normales y células con un solo X. | Fenotipo más variable; puede haber pubertad espontánea parcial o signos más leves. |
| Alteración estructural del X | El cromosoma está presente, pero con una pérdida o reorganización de material genético. | La gravedad depende del segmento perdido y de los genes afectados. |
Hay un matiz que conviene no perder de vista: si aparece material del cromosoma Y, el manejo cambia y debe valorarlo un equipo especializado, porque el riesgo gonadal no es el mismo. Con esta base genética clara, la siguiente pregunta lógica es cuándo empieza a sospecharse en la vida real.
Qué señales la hacen sospechar en la infancia y la adolescencia
La sospecha no nace de un único signo, sino de un conjunto. La talla baja es una de las pistas más frecuentes, pero no basta por sí sola. Si yo tuviera que resumirlo en una frase, diría que el síndrome de Turner suele levantarse cuando hay una combinación de crecimiento por debajo de lo esperado, rasgos físicos concretos y pubertad que no avanza como debería.
En la infancia
- Talla baja o desaceleración del crecimiento respecto a su curva previa.
- Edema de manos o pies al nacer o en los primeros meses.
- Cuello corto o con pliegues, línea de implantación baja del pelo y orejas de implantación baja.
- Alteraciones cardiacas congénitas, sobre todo coartación de aorta o válvula aórtica bicúspide.
- Problemas renales detectados en una ecografía o en estudios por otras causas.
En la pubertad y después
- Ausencia de desarrollo puberal o progreso muy lento.
- Menstruación ausente en la adolescencia por insuficiencia ovárica primaria.
- Infertilidad o dificultades para concebir más adelante.
- Otitis repetidas, pérdida auditiva o cansancio escolar por problemas auditivos no detectados.
- Dificultades concretas en matemáticas, organización visual o atención, aunque la inteligencia global suele ser normal.
El punto práctico es este: una niña con talla baja aislada no siempre tiene esta condición, pero una talla baja persistente junto con pubertad retrasada o malformaciones asociadas sí debe hacer pensar en ella. Y cuando la sospecha ya está sobre la mesa, toca pasar de los signos clínicos a la confirmación genética.
Cómo se confirma el diagnóstico sin pasar por alto un mosaicismo
El estudio de referencia sigue siendo el cariotipo, es decir, el análisis de los cromosomas en sangre. Las guías internacionales más recientes recomiendan analizar al menos 30 metafases como primera prueba, porque así aumenta la probabilidad de detectar mosaicismos. MedlinePlus y las guías clínicas coinciden en algo importante: no basta con ver “un resultado normal” y cerrar el caso si la clínica sigue apuntando a Turner.
En la práctica, el recorrido diagnóstico suele tener varias capas:
- Cariotipo en sangre periférica para confirmar la alteración y describirla con precisión.
- Ampliación del recuento celular si hay sospecha de mosaicismo y el primer resultado no explica bien el cuadro.
- Técnicas complementarias como FISH, PCR o microarray cuando hace falta rapidez, detalle adicional o mayor resolución en ciertos casos.
- Confirmación prenatal, si la sospecha surgió por ecografía o cribado, siempre con una prueba cromosómica diagnóstica y no solo con un test de cribado.
Qué seguimiento y tratamiento cambian el pronóstico
La mejor noticia para una familia es esta: no hay cura única, pero sí muchas intervenciones que mejoran el resultado final. El manejo moderno no se limita a “ver cómo evoluciona”; se adelanta a los problemas más probables. Por eso el enfoque es multidisciplinar y cambia con la edad.
Crecimiento
Si la talla empieza a desviarse, la hormona de crecimiento se valora pronto. Las guías actuales sitúan el inicio temprano alrededor de los 4 a 6 años cuando ya hay retraso de crecimiento, o antes si la velocidad de crecimiento cae con claridad. La respuesta depende de la edad de inicio, de la dosis, de la adherencia y del potencial de crecimiento restante. No prometo una talla “normal”, porque eso sería engañoso; sí puedo decir que iniciar tarde suele dejar menos margen que iniciar a tiempo.
Pubertad y hueso
Cuando la pubertad no arranca sola, la sustitución con estrógenos suele comenzar entre los 11 y 12 años, con ajustes graduales durante los años siguientes. El objetivo no es solo desarrollar caracteres sexuales secundarios; también importa proteger el hueso, el útero y la salud emocional. Empezar demasiado pronto o demasiado tarde puede perjudicar el equilibrio entre crecimiento y maduración, así que aquí el calendario sí importa.
Lee también: Síndrome de Morquio - Guía completa para entenderlo
Corazón, riñones y metabolismo
| Área | Qué se vigila | Por qué importa |
|---|---|---|
| Corazón | Ecocardiografía, aorta y presión arterial; a veces resonancia | Las malformaciones aórticas y la hipertensión son las complicaciones más serias. |
| Riñones | Ecografía renal y función renal según el caso | Las anomalías renales pueden estar presentes desde el nacimiento sin dar síntomas claros. |
| Oído | Revisión audiológica periódica | La pérdida auditiva puede afectar escuela, trabajo y calidad de vida si pasa inadvertida. |
| Tiroides y metabolismo | Función tiroidea, glucosa, lípidos y salud ósea | Hay más riesgo de hipotiroidismo, diabetes tipo 2, osteoporosis y otros problemas metabólicos. |
En esta parte del manejo hay mucha medicina personalizada de verdad: no se trata de seguir una receta fija, sino de ajustar el plan al cariotipo, a los hallazgos cardiacos, al ritmo puberal y a los riesgos de cada paciente. Esa personalización se vuelve todavía más importante cuando entran en juego fertilidad y embarazo.
Fertilidad, menstruación y embarazo sin simplificaciones
Este es uno de los terrenos donde más fácil es generar expectativas incorrectas. Muchas pacientes con Turner presentan insuficiencia ovárica primaria, lo que significa que el ovario deja de funcionar antes de tiempo o nunca llega a hacerlo por completo. Aun así, el panorama no es idéntico en todos los casos: algunas formas en mosaico pueden conservar actividad ovárica parcial y presentar pubertad espontánea, aunque eso no garantiza fertilidad real.
Si existe margen biológico, la preservación de la fertilidad puede valorarse en edades tempranas, pero no es una solución automática ni exenta de debate. Aquí aparece una dimensión bioética clara: decidir cuándo intervenir, qué procedimiento merece la pena, qué riesgos asumir y cómo equilibrar autonomía futura con seguridad presente. En mi opinión, esa conversación debe hacerse con información honesta, sin promesas infladas y sin dramatizar de más.
- La menstruación espontánea puede aparecer en algunos mosaicos, pero no equivale a fertilidad asegurada.
- La ovodonación es una opción reproductiva frecuente, pero no elimina el problema cardiovascular.
- Antes de intentar un embarazo, hace falta valoración por cardiología y medicina materno-fetal.
- El riesgo aórtico durante la gestación es real; una revisión de ASRM sitúa el riesgo de muerte por disección o rotura aórtica en hasta el 1% en mujeres con Turner.
Si tuviera que señalar el error más común, diría que consiste en pensar que el embarazo resuelve o compensa el resto del cuadro. No es así. En esta condición, la seguridad cardiovascular pesa más que el deseo reproductivo y obliga a una evaluación muy seria antes de cualquier intento.
Lo que de verdad marca la diferencia a largo plazo
Si tuviera que resumir el pronóstico en una idea, diría que depende menos del nombre del cariotipo y más de la calidad del seguimiento. Detectar tarde una cardiopatía, normalizar una pubertad ausente o dejar pasar una pérdida auditiva sin tratar suele costar más que cualquier debate semántico sobre el tipo exacto de alteración cromosómica.
- Diagnóstico temprano, porque cada año ganado mejora la planificación hormonal y el control de órganos diana.
- Seguimiento coordinado entre genética, endocrinología, cardiología, nefrología, ginecología y audiología.
- Transición ordenada a la consulta de adultos, para no perder controles cuando termina la etapa pediátrica.
- Apoyo escolar y neuropsicológico si aparecen dificultades visoespaciales, matemáticas o de organización.
- Decisiones reproductivas realistas, basadas en riesgo médico y no en expectativas vagas.
La lectura práctica es sencilla: la monosomía X no se maneja bien con una sola visita ni con una sola especialidad. Cuando el diagnóstico se acompaña de seguimiento riguroso, muchas complicaciones se detectan antes de dar problemas serios. Y ahí está la verdadera diferencia entre conocer el nombre de la alteración y saber cuidar de ella con criterio.