Lo esencial antes de entrar en la técnica
- Detecta variantes en el número de copias del ADN, sobre todo deleciones y duplicaciones pequeñas o medianas.
- No ve bien mutaciones puntuales, reordenamientos equilibrados ni todo lo que depende de la secuencia letra por letra.
- Su resolución suele estar en el rango de 50 a 200 kb, muy por encima del cariotipo clásico, que ronda los 5 Mb.
- Se pide mucho en retraso del desarrollo, discapacidad intelectual, TEA, epilepsia, malformaciones congénitas y anomalías fetales.
- Un resultado útil no depende solo de que salga “positivo”, sino de si la variante encaja con la clínica, es heredada o aparece de novo.
- En prenatal, la interpretación exige más cuidado porque no todas las plataformas ven lo mismo ni responden igual ante mosaicismos o triploidías.
Qué detecta esta técnica y por qué importa
Yo suelo explicarlo de forma sencilla: esta prueba no “lee” el ADN como una secuenciación, sino que compara cuánto material genético hay en cada región. Si falta un fragmento, lo detecta como una deleción; si sobra, como una duplicación. Ese tipo de alteraciones se agrupan dentro de las llamadas CNV o variantes en el número de copias, y muchas de ellas tienen relevancia clínica real.
La utilidad está precisamente ahí. Hay pacientes con un cariotipo aparentemente normal que, sin embargo, presentan microdeleciones o microduplicaciones que explican mejor su cuadro clínico. Por eso esta tecnología cambió la genética médica: no sustituye a todo, pero sí destapa alteraciones submicroscópicas que antes quedaban ocultas. Dicho de otro modo, amplía mucho la capacidad diagnóstica sin obligarte a sospechar una región concreta del genoma desde el principio.
Con esa idea clara, la siguiente pregunta es lógica: ¿qué hace exactamente el laboratorio para comparar dos genomas y traducirlo en un informe clínico?
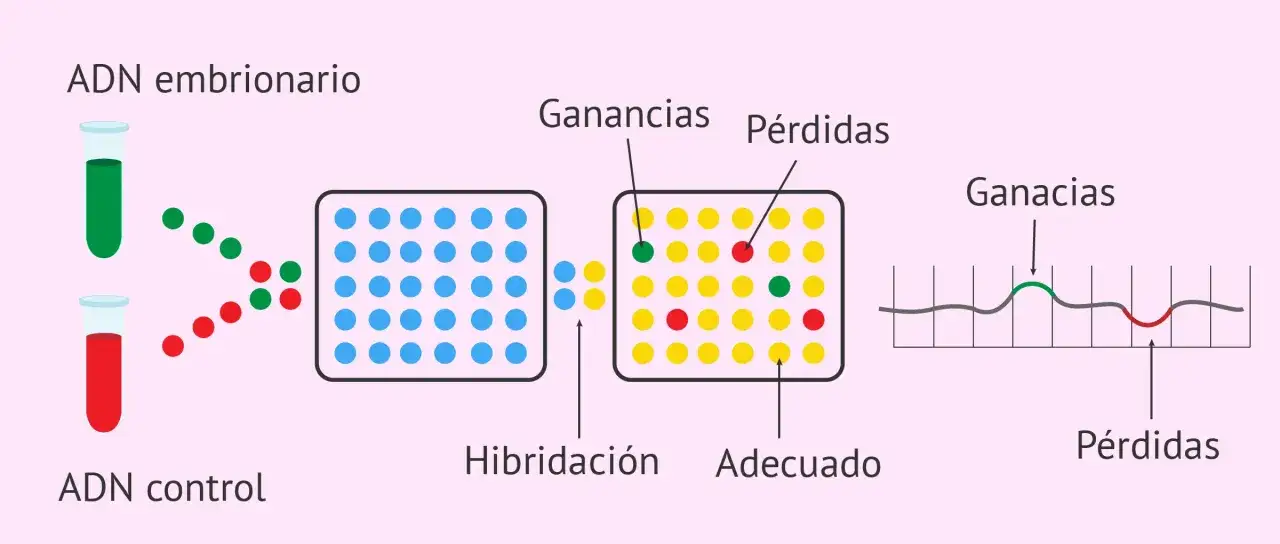
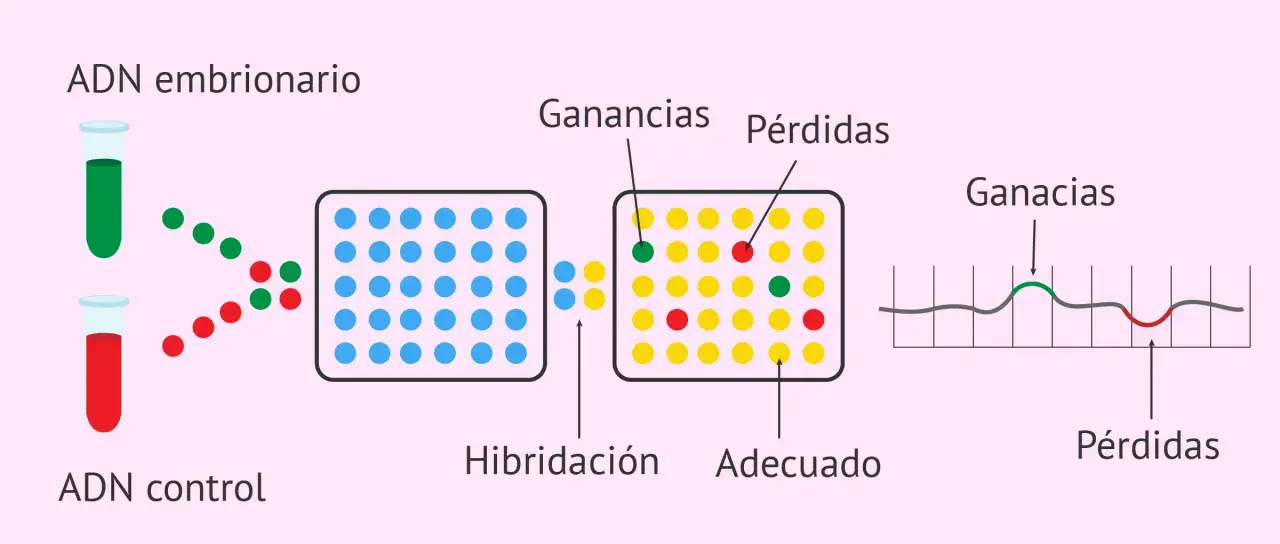
Cómo se hace un estudio de microarrays
El proceso parece complejo, pero en esencia sigue una lógica bastante ordenada. Se extrae ADN de la muestra del paciente y se compara con ADN de referencia marcado con otro color fluorescente. Ambos se hibridan sobre un chip que contiene miles de sondas distribuidas por el genoma. Después, un escáner mide la intensidad de señal en cada punto y el software calcula si hay más o menos material en una región concreta.
La secuencia práctica suele ser esta:
- Extracción de ADN de la muestra biológica.
- Marcaje fluorescente del ADN del paciente y del ADN de referencia.
- Hibridación sobre el microarray con sondas distribuidas por el genoma.
- Lavado, lectura con escáner y medición de señales.
- Análisis bioinformático para identificar pérdidas o ganancias de material genético.
En la práctica, el ensayo suele requerir alrededor de 500 ng de ADN. La resolución depende de la cobertura de la plataforma, pero en microarrays clínicos habituales suele moverse aproximadamente entre 50 y 200 kb; en cariotipo clásico, la detección de alteraciones grandes empieza a rondar los 5 Mb. Cuando la plataforma incorpora SNP además de sondas de aCGH, puede aportar información extra sobre regiones de homocigosidad, uniparentalidad o ciertos cambios de ploidía.
La parte técnica está bien resuelta; lo difícil, de verdad, empieza cuando hay que decidir en qué pacientes merece la pena pedirla.
Cuándo tiene sentido pedirla en España
En España, el uso de microarrays en genética clínica está ya bastante asentado, aunque no de forma idéntica en todos los centros. En prenatal, algunos hospitales la incorporan de forma amplia tras una prueba invasiva; otros la reservan para indicaciones concretas. Esa variabilidad no significa que la técnica sea discutible, sino que el algoritmo clínico depende del circuito asistencial, del tipo de muestra y de la experiencia del laboratorio.
Donde más sentido suele tener es en escenarios en los que sospecho una alteración de número de copias y no quiero quedarme solo con el cariotipo. Las situaciones más típicas son estas:
| Contexto clínico | Qué aporta el estudio | Por qué lo pido |
|---|---|---|
| Retraso del desarrollo o discapacidad intelectual | Busca CNV patogénicas no visibles en cariotipo | Es una de las indicaciones con mayor rendimiento diagnóstico |
| TEA y epilepsia sin causa clara | Puede descubrir deleciones o duplicaciones asociadas al fenotipo | Ayuda a evitar una etiqueta clínica vaga cuando hay una base genética detectable |
| Malformaciones congénitas múltiples o dismorfia | Explora el genoma completo sin tener que elegir una única región | El patrón clínico sugiere una alteración cromosómica desequilibrada |
| Diagnóstico prenatal con anomalías ecográficas | Detecta microdeleciones y microduplicaciones fetales | Añade información cuando el cariotipo o el cribado no bastan |
| Hallazgos familiares o antecedentes de CNV | Permite estudiar herencia y segregación | Ayuda a distinguir una variante de novo de una heredada |
Mi criterio, si lo simplifico, es este: cuanto más sugiera la clínica una alteración de dosis génica, más valor tiene el microarray. Y cuando hay una sospecha clara de mutación puntual en un gen concreto, entonces empiezo a pensar antes en secuenciación. Esa distinción evita peticiones poco rentables y, sobre todo, informes ambiguos.
Una vez indicada la prueba, el siguiente paso no es solo leer el resultado, sino entender qué significa realmente lo que el laboratorio está diciendo.
Cómo leo un informe sin quedarme solo con la palabra “positivo”
Un error muy habitual es fijarse solo en si el informe dice “patogénico” o “normal”. En realidad, el valor de la prueba está en los detalles: tamaño de la región, genes implicados, coordenadas genómicas, herencia familiar y coherencia con el fenotipo del paciente. Yo no me quedo nunca con una sola línea del informe.
Lo más importante es distinguir estas categorías:
- Patogénica o probablemente patogénica: la alteración encaja con enfermedad y tiene peso clínico.
- Variante de significado incierto: hay una diferencia genómica, pero no basta para afirmar que explique el cuadro.
- Benigna o probablemente benigna: no parece tener relevancia clínica.
- Hallazgo heredado frente a de novo: cambia mucho la interpretación, sobre todo si hay un progenitor sano con la misma variante.
La herencia es clave. Una CNV heredada puede ser totalmente inocua, pero también puede tener penetrancia incompleta o expresividad variable. En otras palabras, puede que no todos los portadores muestren el mismo grado de afectación. Por eso, cuando el informe es dudoso, el estudio de los padres suele aclarar más de lo que parece a primera vista.
Además, en genética clínica hay un componente bioético que no conviene trivializar: decidir qué se informa, cómo se informa y con qué nivel de incertidumbre también forma parte del acto médico. Esa es una de las razones por las que el consejo genético pre y post test no es un adorno administrativo, sino una pieza central del proceso.Si ya sabemos cómo se interpreta, la comparación con otras técnicas deja de ser teórica y pasa a ser una decisión práctica.
En qué se diferencia del cariotipo, la FISH, la MLPA y la secuenciación
El error más frecuente es creer que todas las pruebas genéticas hacen lo mismo. No es así. Cada una responde bien a una pregunta distinta, y por eso se complementan mejor de lo que compiten.| Prueba | Qué detecta mejor | Qué puede pasar por alto | Uso típico |
|---|---|---|---|
| Cariotipo | Aneuploidías grandes y reordenamientos cromosómicos amplios | Microdeleciones, microduplicaciones y cambios pequeños | Abortos de repetición, infertilidad, sospecha de translocación equilibrada |
| Microarray aCGH | Pérdidas y ganancias submicroscópicas de ADN | Translocaciones equilibradas, mutaciones puntuales, triploidía y muchos mosaicismos de bajo grado | Retraso del desarrollo, TEA, malformaciones congénitas, anomalías fetales |
| FISH | Una región concreta que ya está sospechada | El resto del genoma | Confirmar o descartar una sospecha puntual |
| MLPA | Deleciones y duplicaciones de genes concretos | Alteraciones genómicas amplias y secuencias puntuales | Cuando el gen diana ya está claro |
| Secuenciación de panel, exoma o genoma | Mutaciones puntuales y pequeñas inserciones o deleciones | Muchos cambios de número de copias si no se analizan de forma específica | Enfermedades monogénicas, fenotipos complejos, casos sin diagnóstico previo |
La lectura práctica es bastante simple: el microarray aporta una visión excelente del número de copias, pero no sustituye a la secuenciación cuando el problema está en la ortografía del gen. Yo lo planteo como una estrategia de capas, no como una carrera entre técnicas. Si el fenotipo sugiere una CNV, empiezo por ahí; si sugiere una enfermedad monogénica, me voy antes a secuenciación.
El matiz importante llega ahora: saber dónde están sus límites evita sobreinterpretaciones y también evita pedirlo donde no va a aportar la respuesta correcta.
Qué conviene revisar antes de firmar el consentimiento
Hay varias limitaciones que conviene tener claras antes de aceptar la prueba. La primera es que el microarray no detecta bien translocaciones equilibradas ni inversiones, porque no cambian la cantidad total de ADN. La segunda es que tampoco sirve para mutaciones puntuales, expansiones de tripletes ni la mayoría de los cambios que solo una secuenciación o una técnica dirigida pueden ver.También hay otros puntos menos intuitivos:
- Triploidía y disomía uniparental: con aCGH puro pueden pasar desapercibidas; un array SNP o una técnica complementaria las resuelve mejor.
- Mosaicismo bajo: alteraciones por debajo de aproximadamente 20-30% de la población celular pueden no detectarse con fiabilidad.
- Variantes de significado incierto: generan incertidumbre clínica y requieren un consentimiento bien explicado.
- Resolución real del chip: en prenatal algunos protocolos trabajan con una resolución global de unos 400 kb, aunque no existe un consenso europeo único.
- Interpretación familiar: sin ADN parental, algunas variantes quedan menos claras de lo que parece.
Antes de aceptar la prueba, yo preguntaría tres cosas muy concretas: qué plataforma se va a usar, si el laboratorio informa variantes inciertas y si la prueba puede necesitar muestras parentales para interpretar bien el resultado. También preguntaría por el tiempo de entrega del informe, que en la práctica suele situarse entre 2 y 6 semanas según la urgencia y el circuito del centro.
Si tuviera que cerrar esta guía con una idea útil, diría que la técnica aporta mucho cuando se pide por la indicación correcta y se interpreta con prudencia clínica. No sustituye al resto del arsenal genético; lo afina. Y precisamente por eso sigue siendo una herramienta central en el diagnóstico prenatal y postnatal cuando lo que buscamos no es una etiqueta rápida, sino una explicación fiable y médicamente útil.