El síndrome de Noonan es una alteración genética que puede afectar al corazón, al crecimiento, a la pubertad, a la coagulación y a varios rasgos físicos desde la infancia. Lo importante no es solo ponerle nombre, sino entender qué órganos conviene vigilar, cómo se confirma la causa y qué decisiones cambian de verdad el pronóstico. En este artículo ordeno la información clínica y genética, con una mirada práctica y útil para familias y para quien quiere entender mejor esta RASopatía.
Lo esencial para orientarse sin perder matices
- Es una RASopatía, es decir, un trastorno ligado a la vía RAS/MAPK, que regula crecimiento y desarrollo celular.
- La expresión es muy variable: puede ir desde rasgos sutiles hasta cardiopatías, talla baja y retraso del desarrollo más visibles.
- El corazón exige atención temprana, porque la cardiopatía congénita es frecuente y condiciona el seguimiento desde el inicio.
- La genética ayuda mucho, pero una prueba negativa no excluye el diagnóstico si el cuadro clínico encaja.
- No existe una cura única: el manejo real es multidisciplinar y se adapta a la edad y a los órganos afectados.
- El consejo genético importa porque cambia el riesgo familiar y las opciones reproductivas cuando se identifica una variante causal.
Qué es el síndrome de Noonan y por qué se clasifica como una RASopatía
Se trata de una RASopatía, es decir, un grupo de enfermedades genéticas relacionadas con la vía RAS/MAPK, que funciona como un sistema de señalización para el crecimiento y la diferenciación celular. Cuando esa vía se altera, el efecto no se queda en un único órgano: puede notarse en el corazón, la estatura, la coagulación, la pubertad, la audición o el desarrollo. En la práctica clínica, eso explica por qué el cuadro es tan variable de una persona a otra.
La frecuencia se estima aproximadamente entre 1 de cada 1.000 y 1 de cada 2.500 nacimientos vivos. No es una enfermedad común, pero tampoco tan rara como para que un pediatra, un cardiólogo infantil o un genetista clínico no la vea alguna vez en consulta.
La causa suele ser una variante patogénica en genes como PTPN11, SOS1, RAF1, RIT1, KRAS, NRAS o LZTR1, entre otros. La herencia es con frecuencia autosómica dominante, aunque muchos casos aparecen de novo, sin antecedentes familiares claros. Cuando uno de los progenitores está afectado, el riesgo para cada hijo es del 50%; en LZTR1 también puede haber herencia recesiva, lo que complica un poco más el mapa familiar.
Yo suelo insistir en una idea: no estamos ante una etiqueta única, sino ante un espectro clínico amplio. Entender eso ayuda a no buscar una “cara típica” que a veces no existe. Con ese marco, la siguiente pregunta útil es cómo se manifiesta realmente en el cuerpo.
Cómo se manifiesta a lo largo de la vida
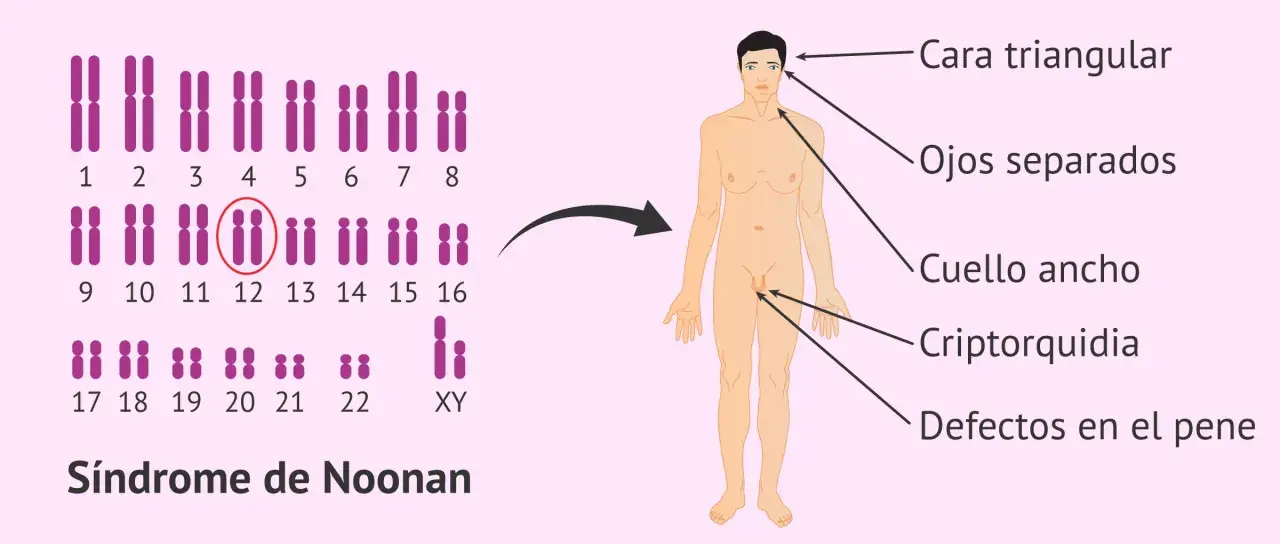
Lo más engañoso de esta enfermedad es que no siempre se presenta igual en todos los momentos. Hay niños con rasgos llamativos desde el nacimiento y otros en los que el diagnóstico se sospecha por la combinación de talla baja, cardiopatía o dificultades de aprendizaje. Los rasgos faciales también pueden hacerse menos evidentes con la edad, así que una foto de recién nacido no basta para entender la historia completa.
En la práctica, hay varios grupos de signos que conviene vigilar:
| Área | Qué puede aparecer | Por qué importa |
|---|---|---|
| Rasgos faciales y cuello | Ojos separados, orejas de implantación baja, cuello corto, pliegue nucal o piel sobrante en la nuca | Orientan el diagnóstico, aunque por sí solos no lo confirman |
| Crecimiento | Talla baja, desaceleración del crecimiento después del nacimiento, retraso de la pubertad | Ayuda a decidir si hace falta valoración endocrina y apoyo nutricional |
| Corazón | Estenosis valvular pulmonar, miocardiopatía hipertrófica, defectos septales u otras cardiopatías congénitas | Es uno de los pilares del pronóstico y del seguimiento temprano |
| Coagulación | Moretones fáciles, epistaxis, sangrado prolongado tras golpes o cirugía | Puede obligar a estudiar la hemostasia antes de procedimientos invasivos |
| Desarrollo y aprendizaje | Retraso de lenguaje, dificultades escolares, atención variable, a veces problemas de conducta o ansiedad | Si se detecta pronto, la intervención educativa cambia mucho el resultado funcional |
| Sexualidad y fertilidad | Criptorquidia en varones, pubertad tardía, fertilidad variable | Importa para el manejo urológico, endocrino y el consejo reproductivo futuro |
| Esqueleto | Tórax en quilla o excavado, escoliosis, complexión torácica poco habitual | Puede requerir seguimiento ortopédico o rehabilitación |
Si hay un dato que merece fijarse es este: la carga médica no siempre se deduce de la apariencia. Muchos niños nacen con peso y talla normales, pero el crecimiento se enlentece después. En paralelo, la cardiopatía congénita aparece en una proporción alta de casos, y la estenosis pulmonar y la miocardiopatía hipertrófica son dos de las formas que más obligan a vigilar. Esa combinación es la que cambia la manera en que se confirma el diagnóstico.
Con esa foto clínica en la mano, el siguiente paso lógico es ordenar las pruebas y no quedarse solo en la impresión externa.
Cómo se confirma el diagnóstico sin sobrerreaccionar a un solo rasgo
La sospecha suele nacer en una combinación de hallazgos: rasgos físicos, antecedentes familiares, cardiopatía, talla baja, problemas de alimentación o retraso del desarrollo. A partir de ahí, lo ideal es no trabajar con una sola prueba aislada, sino con una evaluación escalonada. GeneReviews recuerda que una prueba genética negativa no descarta el cuadro si la clínica es convincente.Yo prefiero pensar el proceso en cuatro capas:
- Sospecha clínica, basada en el patrón completo y no en un rasgo suelto.
- Estudio molecular, normalmente con un panel multigénico de RASopatías o, si hace falta, exoma/genoma.
- Evaluación de órganos diana, sobre todo corazón, crecimiento, desarrollo, audición, visión y coagulación.
- Interpretación genética y familiar, para saber si la variante explica el caso y qué implica para padres y hermanos.
En la práctica, estas son las pruebas que suelen aportar más valor:
| Prueba | Para qué sirve | Qué aporta de forma práctica |
|---|---|---|
| Panel genético multigénico | Busca variantes patogénicas en genes relacionados con la RASopatía | Es la vía más útil cuando el fenotipo ya sugiere el diagnóstico |
| Ecocardiograma y ECG | Detectan cardiopatías estructurales y alteraciones de conducción | Permiten decidir si el seguimiento cardiológico debe ser estrecho desde el inicio |
| Valoración del crecimiento | Compara talla, velocidad de crecimiento y evolución ponderal | Ayuda a distinguir una talla baja constitucional de un patrón patológico |
| Estudio de coagulación | Valora tendencia al sangrado y déficits sutiles de hemostasia | Es especialmente importante antes de cirugía o si hay moretones o epistaxis |
| Audición y visión | Busca hipoacusia, estrabismo, errores de refracción o ptosis | Puede explicar parte de las dificultades escolares o del lenguaje |
| Desarrollo y lenguaje | Evalúa motricidad, cognición, comunicación y conducta | Define si hace falta estimulación temprana o apoyo educativo |
| Ecografía renal y exploración genitourinaria | Detecta anomalías renales o criptorquidia | Orientan derivación a urología o nefrología cuando procede |
Un detalle práctico que suele pasar desapercibido: no conviene esperar a tener la genética cerrada para empezar el resto del circuito. Si el niño tiene soplo, mala ganancia ponderal o sospecha de retraso del desarrollo, esas derivaciones deben moverse ya. Eso evita perder meses valiosos y, además, ayuda a no confundir una RASopatía con otros cuadros parecidos.
Justamente por eso merece la pena detenerse un momento en el diagnóstico diferencial, porque ahí es donde más errores de interpretación veo en la práctica.
Con qué otros cuadros se confunde con facilidad
No todo paciente con cuello corto, talla baja y cardiopatía tiene el mismo diagnóstico. La frontera entre síndromes con solapamiento fenotípico puede ser borrosa, y eso importa porque el pronóstico, el consejo genético y el seguimiento no son idénticos. El problema no es solo poner un nombre incorrecto, sino organizar mal la vigilancia desde el principio.
| Cuadro parecido | Qué ayuda a distinguirlo | Por qué no conviene confundirlo |
|---|---|---|
| Síndrome de Turner | Afecta casi siempre a niñas y depende de una alteración cromosómica sexual | El enfoque genético y reproductivo es distinto |
| Síndrome cardiofaciocutáneo | Suele dar más afectación cutánea, capilar y neurodesarrollo más marcado | Comparte vía molecular, pero el patrón clínico suele ser más intenso |
| Síndrome de Costello | Más problemas de alimentación, facies más tosca y mayor carga dermatológica | Puede implicar un riesgo oncológico y evolutivo diferente |
| Neurofibromatosis tipo 1 | Aparición de manchas café con leche, neurofibromas o pecas axilares | La orientación diagnóstica y el seguimiento cambian bastante |
| Síndrome de Aarskog | Hay solapamiento en rasgos faciales y talla baja, pero no el mismo patrón genético | El consejo familiar y la interpretación clínica son diferentes |
La clave, si tuviera que resumirla, es esta: no basta con ver una “cara de RASopatía”. Hace falta integrar corazón, crecimiento, coagulación, desarrollo y genética. Cuando esa lectura se hace bien, el siguiente paso ya no es etiquetar más, sino tratar y seguir mejor.
Y ahí es donde el manejo multidisciplinar deja de ser una idea bonita para convertirse en una necesidad real.
Qué tratamiento y seguimiento suelen marcar la diferencia
Un consenso reciente en JAMA Network Open insiste en algo que, en realidad, ya se veía en la práctica: el manejo debe ser multidisciplinar, longitudinal y adaptado a cada paciente. No hay un tratamiento único que resuelva todo, pero sí hay una secuencia de decisiones que cambia mucho el resultado funcional y médico.
| Área | Qué se suele hacer | Comentario práctico |
|---|---|---|
| Cardiología | Ecocardiografía, ECG y tratamiento específico si hay estenosis pulmonar, miocardiopatía hipertrófica u otras cardiopatías | Si el estudio inicial es normal, en la infancia suele mantenerse al menos una revisión anual hasta los 5 años y luego cada 5 años o antes si hay síntomas |
| Endocrinología | Seguimiento de talla y velocidad de crecimiento, edad ósea, función tiroidea y valoración de hormona del crecimiento en casos seleccionados | La hormona del crecimiento puede considerarse en talla baja significativa, pero la indicación es individual y no automática |
| Neurodesarrollo | Estimulación temprana, logopedia, apoyo psicopedagógico y seguimiento de atención, ansiedad o TEA cuando procede | Empezar pronto suele rendir más que esperar a que el retraso “se compense solo” |
| Hematología | Estudio de sangrado, recuento celular y pruebas de coagulación si hay antecedentes o antes de cirugía | Es una de las piezas que más se olvida y una de las que más evita problemas en quirófano |
| Nutrición y digestivo | Apoyo en dificultades de alimentación, disfagia o mala ganancia ponderal; a veces sonda o gastrostomía | En lactantes con cardiopatía o cardiomiopatía, el soporte nutricional puede ser decisivo |
| ORL, audiología y oftalmología | Control de audición y visión, corrección de estrabismo, ptosis o hipoacusia | Una parte de las dificultades escolares mejora mucho cuando se corrige esto a tiempo |
| Urología | Valoración de criptorquidia y otras anomalías genitourinarias | Importa tanto por fertilidad futura como por salud testicular |
Hay una regla práctica que yo consideraría casi obligatoria: antes de cualquier cirugía o procedimiento invasivo, revisar el antecedente hemorrágico y, si procede, ampliar el estudio de coagulación. En este síndrome, los estudios básicos pueden no contar toda la historia. También conviene no subestimar la alimentación en lactantes, porque una mala ganancia de peso puede ser la primera pista de que el problema es más que “un niño que come poco”.
Una vez ordenado el seguimiento, la conversación con la familia cambia de nivel. Ya no se trata solo de tratar síntomas, sino de entender el riesgo familiar y las opciones reproductivas.
Qué implica para la familia, el embarazo y el consejo genético
Este es el punto donde la genética y la bioética se tocan de forma más clara. Cuando una variante causal está identificada, la enfermedad suele comportarse como autosómica dominante, así que cada hijo de una persona afectada tiene un 50% de probabilidad de heredar la variante. Si el caso es de novo y ninguno de los padres la porta, el riesgo para hermanos suele ser bajo, aunque no es exactamente cero por la posibilidad de mosaicismo germinal.
Si la variante ya se conoce en la familia, el laboratorio puede ofrecer diagnóstico prenatal y también diagnóstico genético preimplantacional. Técnicamente es posible, pero conviene entender sus límites: saber que el feto porta la variante no permite predecir con precisión si el curso será leve, moderado o más complejo. En otras palabras, el diagnóstico molecular confirma la causa, pero no cierra del todo el pronóstico.
También hay que ser prudentes con la ecografía prenatal. Puede mostrar hallazgos sugerentes, como aumento de la translucencia nucal, edema, derrames o ciertas cardiopatías, pero esos datos no son específicos. Sirven para sospechar, no para cerrar por sí solos el caso. Por eso, en consejo genético yo insistiría en una frase sencilla: más información no siempre significa más certeza, y la decisión reproductiva debe asumir ese margen de incertidumbre.
- Si hay un progenitor afectado, el riesgo de transmisión es del 50%.
- Si la variante no está en los padres, el riesgo para hermanos baja mucho, pero no desaparece por completo.
- Si se plantea un embarazo, conviene hablar antes de opciones diagnósticas y de sus límites.
- Si la familia necesita una decisión reproductiva, la parte técnica y la parte emocional deben ir juntas, no por separado.
Con todo eso sobre la mesa, lo que más importa a largo plazo ya no es solo la causa genética, sino la manera en que se organiza el seguimiento cotidiano.
Lo que realmente cambia el pronóstico a largo plazo
Si tengo que quedarme con una idea útil para familias y clínicos, es esta: el pronóstico mejora más por un seguimiento bien coordinado que por una única intervención aislada. Detectar pronto una cardiopatía, vigilar la coagulación antes de una cirugía, sostener la nutrición en los primeros años y activar apoyo del lenguaje cuando hace falta cambia más que muchas medidas tardías y fragmentadas.
- Cardiología desde el inicio, aunque el niño parezca estable.
- Seguimiento del crecimiento en cada visita, no solo cuando ya hay una talla baja muy marcada.
- Apoyo del desarrollo y del lenguaje si aparecen retrasos o dificultades escolares.
- Revisión auditiva y visual periódica, porque a veces ahí se esconde parte del problema funcional.
- Consejo genético realista, con información útil y sin prometer certezas que la biología no ofrece.
El síndrome de Noonan se maneja mejor cuando genética, cardiología, endocrinología, hematología y desarrollo infantil trabajan como un solo circuito. Esa coordinación es la que reduce complicaciones y la que permite que cada paciente reciba un plan ajustado a su forma concreta de presentar la enfermedad.