Las enfermedades genéticas raras no forman un bloque uniforme: detrás de esa etiqueta hay cuadros muy distintos, desde alteraciones de un solo gen hasta cambios cromosómicos o mecanismos más complejos que afectan al desarrollo, al metabolismo o al sistema nervioso. En este artículo explico qué las caracteriza, por qué el diagnóstico suele tardar tanto, qué tipos existen, qué pruebas se usan hoy y qué cambia para la familia cuando por fin llega una respuesta útil.
Las claves que conviene tener claras desde el principio
- En Europa, una enfermedad se considera rara cuando afecta a menos de 5 personas por cada 10.000.
- La gran mayoría de los trastornos raros tienen base genética, pero eso no significa siempre que se hereden de forma directa.
- Los signos suelen ser multisistémicos, progresivos o muy variables, y a menudo empiezan en la infancia.
- El diagnóstico ha mejorado mucho con paneles, exoma y genoma, pero un resultado negativo no cierra el caso.
- En España, la medicina genómica está acortando el recorrido diagnóstico, aunque la odisea sigue siendo frecuente.
- El consejo genético importa tanto como el nombre de la enfermedad, porque cambia decisiones médicas y familiares.
Qué hace rara a una enfermedad de base genética
La palabra “rara” describe sobre todo su frecuencia, no su importancia clínica. En el marco europeo, una patología entra en esa categoría cuando afecta a menos de 1 de cada 2.000 personas, pero en conjunto representan un problema enorme de salud pública. Lo que más llama la atención es la paradoja: cada enfermedad aislada es poco frecuente, pero el conjunto afecta a millones de personas.
Cuando hablamos de trastornos de base genética poco frecuentes, la rareza no suele venir sola. Casi siempre se acompaña de heterogeneidad clínica, dificultad diagnóstica y una gran dispersión de casos, lo que complica reunir experiencia suficiente en un único centro. Yo suelo fijarme en eso antes que en el nombre exacto: lo que importa es el patrón, no solo la etiqueta.
Herencia no siempre significa antecedentes familiares
Este es uno de los errores más comunes. Una enfermedad puede ser genética y, aun así, aparecer “de novo”, es decir, por una variante nueva en el paciente, sin historia familiar previa. O puede heredarse de forma recesiva, donde ambos progenitores son portadores sanos, o ligada al cromosoma X, o incluso por alteraciones mitocondriales que siguen otro patrón.
En la práctica, eso significa que la ausencia de casos en la familia no descarta un origen genético. También significa que el árbol familiar, bien hecho, vale mucho más de lo que parece al principio. Y ese matiz lleva directamente a las señales clínicas que más ayudan a sospecharlas.
Lee también: Anemia falciforme - Guía completa: causas, síntomas y tratamiento
Por qué la rareza complica el recorrido asistencial
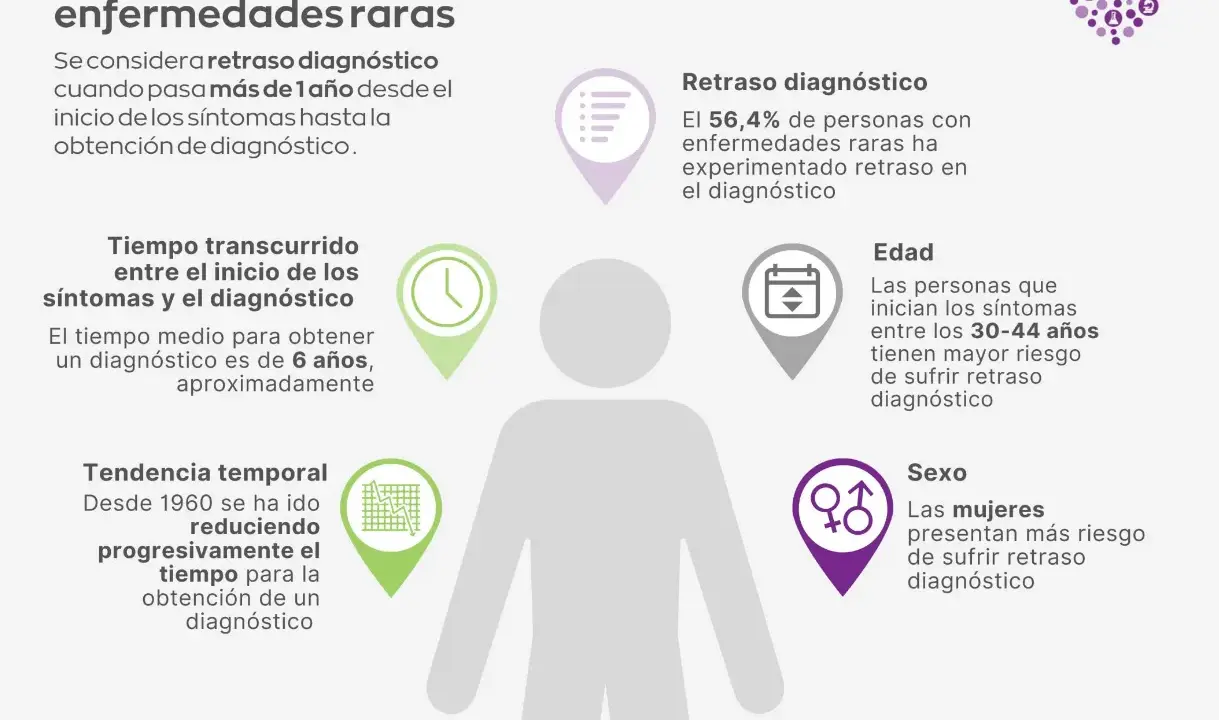
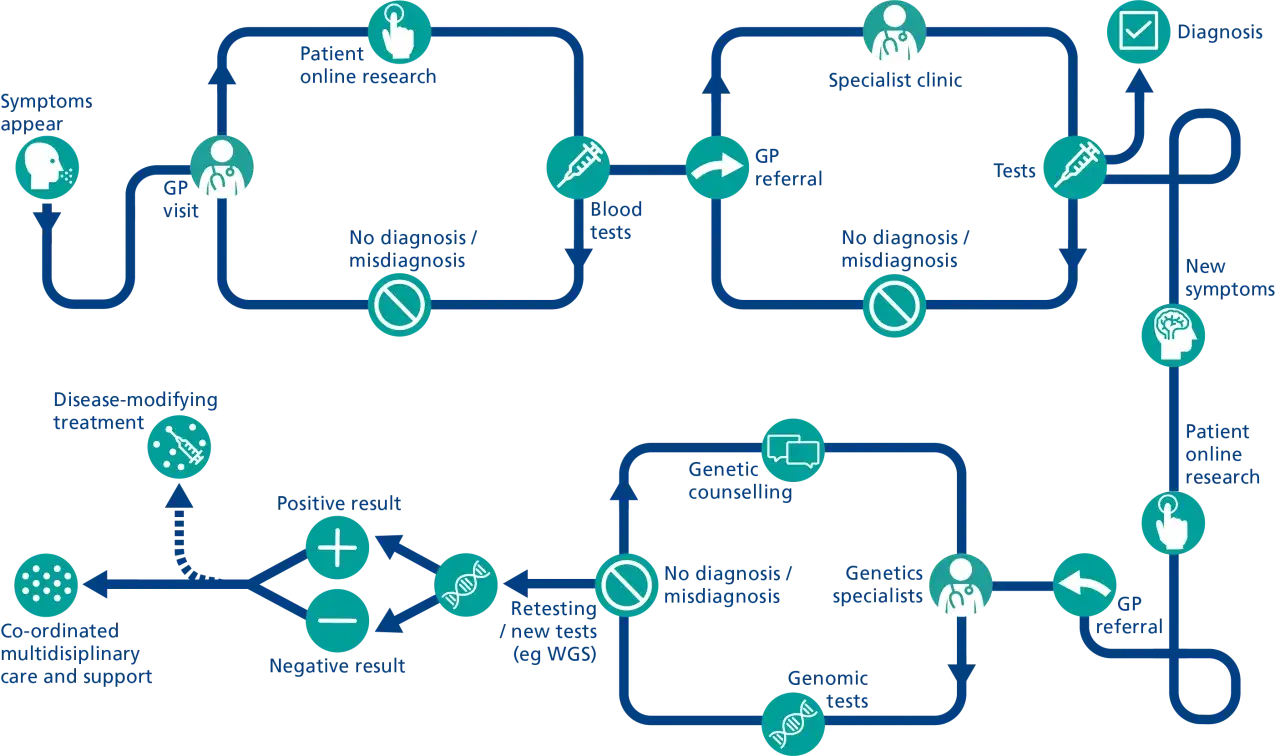
La combinación de baja frecuencia, síntomas poco específicos y variabilidad entre pacientes hace que muchos casos pasen por varias consultas antes de encajar. El Ministerio de Sanidad ha señalado que gran parte de estas enfermedades tienen base genética y que, durante años, el tiempo hasta el diagnóstico en España ha sido demasiado largo. Esa demora no es un detalle administrativo: cambia el pronóstico, el acceso a terapias y la carga emocional de la familia.
Por eso, cuando un cuadro no encaja bien en una especialidad concreta, conviene pensar en una visión transversal. Esa forma de mirar prepara el terreno para reconocer los signos que suelen repetirse.
Las señales que suelen repetirse
No existe una lista universal de síntomas, pero sí un grupo de pistas que, juntas, deberían levantar sospecha. La clave no es que haya un síntoma “exótico”, sino que varias piezas pequeñas no expliquen bien el cuadro completo. Yo suelo resumirlo así: cuando una enfermedad toca varios órganos, avanza sin una lógica clara o aparece muy pronto, merece una mirada genética seria.
- Afectación multisistémica: por ejemplo, neurológica y muscular al mismo tiempo, o digestiva y neurológica, o cardiaca y esquelética.
- Inicio temprano: muchas debutan en la infancia, aunque algunas no se manifiestan hasta la edad adulta.
- Evolución progresiva o por brotes: el paciente mejora y empeora sin una explicación sencilla.
- Retraso del desarrollo o discapacidad intelectual: sobre todo si se acompaña de epilepsia, hipotonía o rasgos dismórficos.
- Historia familiar sugestiva: varios afectados, consanguinidad, abortos recurrentes o patrones de transmisión llamativos.
- Variabilidad clínica: dos personas con la misma variante pueden tener síntomas muy distintos.
En genética clínica, a esa combinación de síntomas y signos se le llama fenotipo, es decir, la forma en que una alteración biológica se expresa en la persona. También conviene recordar dos conceptos que cambian mucho la interpretación: penetrancia, que indica cuántos portadores manifiestan la enfermedad, y expresividad, que mide cuánto varía la intensidad entre pacientes.
Si el fenotipo es poco claro, el siguiente paso no es adivinar un nombre, sino pensar en el mecanismo. Y ahí es donde los ejemplos ayudan a ordenar el mapa.
Tipos y ejemplos que ayudan a entenderlas
Prefiero ordenar estos cuadros por mecanismo, porque así se entiende mejor qué busca el laboratorio y por qué algunos tests sirven más que otros. No me interesa acumular nombres; me interesa que el lector vea el patrón clínico detrás de cada uno.| Tipo | Qué ocurre | Ejemplo | Qué enseña al clínico |
|---|---|---|---|
| Monogénica | Una variante en un solo gen altera una proteína o su función. | Fibrosis quística, atrofia muscular espinal | La confirmación molecular puede abrir tratamientos dirigidos y cambia el consejo familiar. |
| Cromosómica o por número de copias | Hay pérdidas, duplicaciones o reordenamientos grandes de material genético. | Deleción 22q11.2, síndrome de Williams | Las pruebas de secuenciación no siempre bastan; a veces hacen falta microarrays o técnicas estructurales. |
| Mitocondrial | El problema afecta al ADN mitocondrial o a genes nucleares ligados a la función energética. | Neuropatía óptica hereditaria de Leber, síndrome de Leigh | La herencia y la expresión clínica siguen reglas distintas de la herencia mendeliana clásica. |
| Expansión de repeticiones | Un fragmento de ADN se repite demasiadas veces y altera la función del gen. | Enfermedad de Huntington, algunas ataxias hereditarias | La edad de inicio y la gravedad pueden depender del tamaño de la expansión. |
| Impronta genética o epigenética | Importa de qué progenitor procede el material genético o cómo está regulado. | Prader-Willi, Angelman | Explican por qué dos alteraciones parecidas pueden dar cuadros clínicos muy distintos. |
Con ese mapa en mente, la siguiente cuestión lógica es cómo se diagnostican hoy de forma más eficaz, especialmente en España.
Cómo se diagnostican hoy en España
Yo separo siempre dos fases: sospecha clínica y confirmación genética. Confundirlas lleva a pruebas mal elegidas, a resultados poco interpretables y a falsas sensaciones de cierre. La primera parte sigue estando en la consulta: historia clínica detallada, exploración física fina, árbol familiar y una descripción clara del fenotipo. La segunda entra en juego cuando ya se sabe qué buscar.
Hoy se usan distintas herramientas según el caso: paneles de genes, microarrays, secuenciación del exoma, secuenciación del genoma completo y, en algunos escenarios, técnicas capaces de detectar variantes estructurales difíciles de ver con métodos clásicos. La elección del test importa tanto como el test en sí. Un panel muy estrecho puede pasar por alto la causa; un genoma completo mal interpretado puede generar ruido innecesario.
| Prueba | Cuándo suele ayudar | Qué aporta |
|---|---|---|
| Panel dirigido | Cuando el fenotipo sugiere una familia concreta de genes. | Es rápido, más acotado y reduce hallazgos irrelevantes. |
| Exoma | Cuando el cuadro es compatible con base genética, pero el gen exacto no está claro. | Amplía mucho la búsqueda en regiones codificantes. |
| Genoma completo | Cuando el exoma es negativo o se sospechan variantes complejas. | Permite explorar más allá de las regiones codificantes y revisar mejor la arquitectura global del ADN. |
| Estudios estructurales avanzados | Cuando se sospechan reordenamientos, inserciones o deleciones complejas. | Detecta cambios que la secuenciación clásica puede pasar por alto. |
En España, la red pública está avanzando hacia una oferta más homogénea de pruebas genéticas, y eso es buena noticia para pacientes con cuadros complejos. Además, el ISCIII ha mostrado que el reanálisis de exomas añadió un 12% de diagnósticos nuevos y el estudio del genoma completo otro 18% en casos sin respuesta previa. Esa cifra resume bien una idea clave: un resultado negativo hoy no siempre es definitivo.
También conviene tener presente que el retraso diagnóstico sigue siendo una realidad. Distintas series españolas han descrito medias cercanas a cuatro años, y en una parte relevante de las familias el recorrido supera los diez. En la práctica, eso obliga a insistir en la reevaluación cuando el cuadro evoluciona, cuando aparecen nuevos síntomas o cuando cambian las herramientas disponibles.
El siguiente paso natural es preguntarse qué cambia cuando por fin se conoce el mecanismo. Y la respuesta es: bastante más de lo que parece.
Qué cambia en el tratamiento cuando conoces el gen
Encontrar la causa no equivale siempre a curar, pero sí cambia el juego. Permite decidir mejor el seguimiento, anticipar complicaciones, ajustar rehabilitación, valorar terapias dirigidas y abrir la puerta a ensayos clínicos. En algunas enfermedades, el diagnóstico molecular llega a tiempo para marcar una diferencia real en la evolución.Hay tres grandes niveles de intervención. El primero es el tratamiento sintomático y de soporte: control de crisis epilépticas, fisioterapia, nutrición, soporte respiratorio, rehabilitación, cardiología o neurología según el órgano afectado. El segundo es el tratamiento dirigido al mecanismo, que ya existe en algunas enfermedades concretas, como terapias enzimáticas, moduladores de proteínas o fármacos diseñados para una variante específica. El tercero es la terapia génica o basada en ARN, todavía limitada a un número reducido de casos, pero cada vez más relevante.
Lo importante aquí es no vender falsas expectativas. No todas las patologías tienen tratamiento causal y no todas responden igual aunque exista una terapia aprobada. El estadio clínico, la edad de inicio y el tipo exacto de variante influyen mucho. A veces el mayor beneficio llega por un detalle poco glamuroso pero decisivo: haber llegado antes al diagnóstico.
Si la parte médica cambia, la parte familiar cambia todavía más. Y ahí entra el consejo genético.
Lo que la familia necesita saber antes de tomar decisiones
Cuando hay una variante patogénica confirmada, la familia deja de moverse a ciegas. Ese es el valor del consejo genético: explicar qué significa el hallazgo, cómo se transmite, qué riesgo real existe para otros miembros y qué opciones reproductivas o preventivas son razonables en ese caso concreto. No es un trámite accesorio; es una pieza central del proceso.
- Autosómica dominante: basta una copia alterada para que exista riesgo de enfermedad; cada hijo suele tener un 50% de probabilidad de heredarla.
- Autosómica recesiva: ambos progenitores suelen ser portadores; el riesgo por embarazo puede ser del 25% para un hijo afectado.
- Ligada al cromosoma X: el patrón de riesgo cambia según el sexo biológico y el gen implicado.
- Mitochondrial: la transmisión suele seguir la línea materna.
También pueden plantearse estudio de portadores, diagnóstico prenatal o diagnóstico genético preimplantacional, según el caso y el marco asistencial disponible. Aquí suelo ser muy prudente: no todas las parejas necesitan las mismas opciones, y no toda opción disponible es la más adecuada. La decisión útil es la que encaja con la variante, la historia clínica y los valores de la familia.
Hay otro punto que casi siempre aparece tarde, y no debería: el impacto emocional. Recibir una respuesta genética puede aliviar la incertidumbre, pero también abre preguntas difíciles sobre hermanos, hijos, culpa, planificación y seguimiento a largo plazo. Si ese componente se gestiona bien, el beneficio del diagnóstico es mucho mayor.
Lo que conviene recordar para no perder tiempo valioso
Si algo resume bien este tema es que la rareza no reduce el impacto, solo hace más difícil reconocerlo a tiempo. Las enfermedades hereditarias poco frecuentes suelen ser complejas, pero no misteriosas: dejan pistas clínicas, familiares y moleculares que hoy pueden rastrearse mucho mejor que hace unos años.
Mi recomendación práctica es simple: cuando un cuadro es multisistémico, empieza pronto, no encaja bien o deja demasiadas piezas sueltas, merece una valoración genética completa y, si hace falta, una reevaluación posterior. Esa combinación de sospecha clínica, tecnología adecuada y consejo genético es la que más cambia la historia real del paciente.
En la consulta, casi siempre merece la pena hacerse la misma pregunta una vez más: si esto no es casualidad, ¿qué dato me falta para entenderlo mejor?