Lo esencial para orientarse sin perder tiempo
- El problema principal no está en el músculo, sino en las neuronas motoras de la médula espinal.
- La mayoría de los casos se relaciona con alteraciones en SMN1; SMN2 suele modular la gravedad.
- La enfermedad se presenta en un espectro amplio, desde formas muy precoces y graves hasta cuadros leves en adultos.
- En España, el cribado neonatal del SNS ya incorpora esta patología, algo clave porque el tratamiento precoz cambia el pronóstico.
- Los fármacos actuales no curan, pero sí pueden frenar la evolución si se inician pronto y se combinan con soporte respiratorio, nutricional y rehabilitación.
- El consejo genético es parte del manejo, no un añadido opcional, porque ayuda a estimar el riesgo familiar y a planificar embarazos.
Qué es la AME y por qué afecta al movimiento
Cuando explico esta enfermedad, empiezo por una idea simple: no es un problema muscular primario, sino un fallo de las neuronas motoras. Esas células envían la orden de movimiento al músculo; si se pierden o funcionan mal, el músculo se debilita y se va afinando con el tiempo. La mayoría de los casos se asocia a alteraciones en SMN1, un gen necesario para fabricar una proteína esencial para la supervivencia de esas neuronas.
También importa SMN2, un gen muy parecido que puede compensar parcialmente la falta de SMN1. No resuelve el problema por completo, pero sí ayuda a explicar por qué unas personas tienen una forma más grave y otras una evolución más lenta. En la práctica, la AME no se entiende como una sola enfermedad rígida, sino como un espectro con distinto grado de debilidad, distinta edad de inicio y distinta velocidad de progresión.
La herencia suele ser autosómica recesiva: una persona puede ser portadora sin síntomas y transmitir la variante genética sin saberlo. Esa es una de las razones por las que el diagnóstico genético y el asesoramiento familiar tienen tanto peso. La siguiente pregunta lógica es cómo se manifiesta, porque aquí la edad de inicio cambia mucho la historia clínica.
Cómo se reconoce según la edad de inicio
Mi forma de ordenarlo es por el momento en que aparecen los síntomas. No porque la etiqueta lo explique todo, sino porque ayuda a entender la gravedad inicial, el riesgo respiratorio y el grado de apoyo que suele necesitar cada paciente.
| Tipo | Inicio típico | Señales frecuentes | Curso habitual |
|---|---|---|---|
| Tipo 1 | Antes de los 6 meses | Hipotonía, dificultad para tragar, debilidad marcada, escaso control de tronco y problemas respiratorios | Es la forma más grave; sin tratamiento, el riesgo de complicaciones tempranas es alto |
| Tipo 2 | Entre los 6 y los 18 meses | Puede sentarse sin apoyo, pero no suele ponerse de pie ni caminar sin ayuda; son comunes la escoliosis y las infecciones respiratorias | Evolución intermedia, con necesidad variable de ayuda motora y respiratoria |
| Tipo 3 | Después de los 18 meses | Camina sola al inicio, pero le cuesta correr, levantarse de una silla o subir escaleras | Suele ser más leve, aunque puede haber pérdida progresiva de fuerza y limitaciones funcionales |
| Tipo 4 | Edad adulta | Debilidad leve o moderada, temblor y, a veces, molestias respiratorias leves | La progresión suele ser lenta y la expectativa de vida suele ser normal |
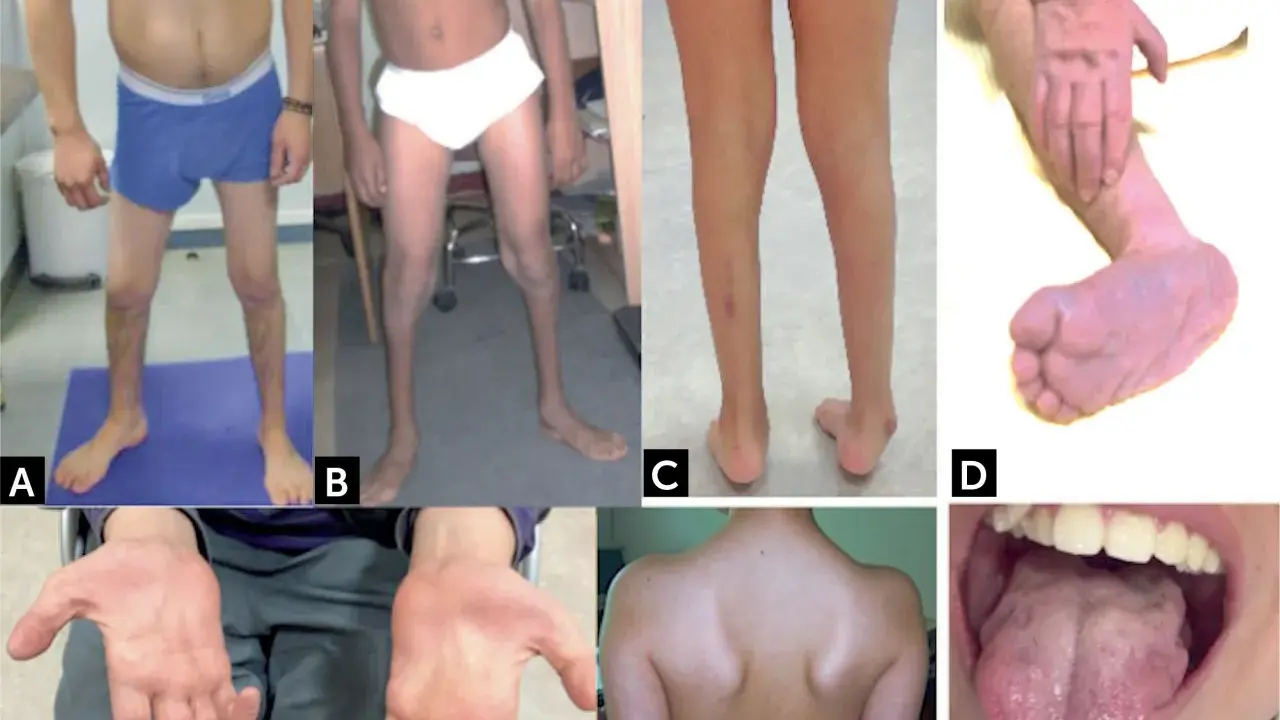
En consulta, los signos que más me hacen pensar en este diagnóstico son la hipotonía o tono muscular bajo, la arreflexia o ausencia de reflejos, la debilidad proximal y la pérdida de habilidades que antes ya estaban adquiridas. No todos los pacientes llegan igual, pero hay un patrón que se repite: el problema no suele ser aislado en una sola extremidad, sino más difuso y simétrico. Con ese mapa en mente, el siguiente paso es confirmar la sospecha sin perder tiempo.
Cómo se confirma el diagnóstico y por qué el tiempo importa
La clave diagnóstica es la genética. El estudio suele buscar primero la alteración en SMN1 y, si hace falta, completar con técnicas más finas para no dejar casos sin explicación. La electromiografía y los estudios de conducción nerviosa pueden ayudar, sobre todo si el cuadro no está claro, pero hoy el diagnóstico definitivo se apoya en la prueba molecular. En algunos casos, el número de copias de SMN2 ayuda a estimar la gravedad probable, aunque no sustituye al diagnóstico.
En España, el Ministerio de Sanidad ha incorporado esta enfermedad al cribado neonatal del SNS, algo muy relevante porque la ventana terapéutica es más favorable antes de que el daño neuromuscular sea irreversible. Eso sí, conviene no confundir cribado con diagnóstico: una prueba del talón alterada no cierra el caso, solo acelera la confirmación y la derivación a una unidad especializada.
Si hay antecedentes familiares, también pueden valorarse pruebas prenatales. Y aquí me parece importante ser preciso: cuando hay una variante en ambos progenitores, cada embarazo tiene un 25% de probabilidad de estar afectado, un 50% de probabilidad de que el hijo o hija sea portador y un 25% de probabilidad de no heredar la alteración. Ese dato no sirve para alarmar, sino para entender por qué el consejo genético no es un trámite burocrático, sino parte de la atención clínica.
La lógica es clara: cuanto antes se confirma, antes se decide el tratamiento y antes se evita pérdida funcional. Y eso enlaza directamente con la pregunta más importante para la mayoría de las familias: qué opciones reales hay hoy.
Qué tratamientos modifican hoy la evolución
MedlinePlus recuerda que no existe una cura, pero sí tratamientos capaces de controlar síntomas, prevenir complicaciones y, en muchos casos, cambiar de forma notable la evolución. Yo suelo dividirlos en dos grupos: los que actúan sobre la causa biológica y los que sostienen la función del paciente mientras el sistema nervioso se protege o se recupera parcialmente.
| Tratamiento | Cómo se administra | Qué aporta | Qué hay que tener en cuenta |
|---|---|---|---|
| Nusinersen | Vía intratecal | Modula el procesamiento de SMN2 para aumentar proteína funcional | Requiere punciones lumbares periódicas |
| Risdiplam | Vía oral diaria | Actúa sobre SMN2 y tiene alcance sistémico | La adherencia diaria importa mucho |
| Onasemnogene abeparvovec | Terapia génica de una sola administración | Introduce una copia funcional del gen implicado | La selección del paciente y la monitorización hepática son decisivas |
La parte menos visible, pero a menudo la más determinante en la vida diaria, es el soporte multidisciplinar. Aquí entran la fisioterapia, la terapia ocupacional, el control de la postura, las órtesis, la nutrición, el manejo de la disfagia y el soporte respiratorio cuando hace falta. En otras palabras: aunque el tratamiento modificador de la enfermedad cambie la curva clínica, el resultado real depende mucho de que el paciente no llegue tarde al circuito de cuidados.
Yo suelo insistir en un matiz que a veces se pasa por alto: los tratamientos funcionan mejor antes de la pérdida motora marcada, no después. Eso no significa que no haya beneficio en fases más avanzadas, pero sí que las expectativas deben ser realistas. Cuanto más tiempo lleva la neurona dañada, más difícil es revertir por completo el daño. Y eso lleva a otra pregunta incómoda pero necesaria: qué significa esta enfermedad para la familia.
Qué cambia para la familia y el consejo genético
Esta es una enfermedad que no solo se atiende con neurología o pediatría, sino también con genética clínica. Si una persona afectada tiene hermanos, hijos o desea tener descendencia, el consejo genético ayuda a aclarar riesgos, opciones de estudio y decisiones reproductivas. No se trata de medicalizar la vida familiar, sino de dar información clara para decidir con menos incertidumbre.
En la práctica, el estudio de portadores puede ser útil en la pareja reproductiva y en familiares cercanos, sobre todo cuando ya hay un caso confirmado. También puede valorarse el diagnóstico prenatal si existe una variante familiar conocida. La lógica es sencilla: si se identifica la alteración concreta, se puede orientar mejor el seguimiento y la planificación.
Hay otro punto que considero importante: los padres suelen vivir con culpa injustificada, como si la enfermedad se debiera a algo que hicieron. No es así. La mayoría de las veces hablamos de portadores sanos que desconocían su situación genética. Entender esto cambia mucho la conversación clínica, porque desplaza el foco desde la culpa hacia la información útil.
Y esa información útil no acaba en el diagnóstico familiar; sigue en el seguimiento a largo plazo, donde de verdad se ve si el sistema asistencial acompaña bien al paciente.
Lo que más cambia el pronóstico cuando se actúa a tiempo
Si tuviera que resumir la diferencia entre una evolución peor y una mejor, no diría solo “más tratamiento”, sino más tiempo ganado antes del daño. La combinación que más cambia el pronóstico es clara: detección temprana, confirmación genética rápida, inicio precoz del tratamiento modificador y seguimiento multidisciplinar constante.
- La respiración debe vigilarse incluso cuando el problema motor parece el más visible.
- La nutrición importa tanto como la fuerza, porque tragar mal desgasta y empeora el estado general.
- La columna y las articulaciones necesitan seguimiento, ya que la escoliosis y las contracturas aparecen con facilidad.
- Las infecciones respiratorias pueden descompensar a un paciente que, en apariencia, estaba estable.
- La transición a la atención del adulto no debe improvisarse en los casos que llegan a la vida adulta.
La lectura final es bastante clara: esta enfermedad ya no se mira con la resignación de hace unos años. Hoy hay herramientas para diagnosticar antes, tratar antes y sostener mejor a cada paciente, pero el beneficio depende de no retrasar la derivación ni subestimar señales como la hipotonía, la debilidad proximal o los problemas para alimentarse. Cuando ese circuito funciona, el pronóstico cambia de forma real; cuando se retrasa, se pierde parte de la ventaja terapéutica. Y en una patología así, ese margen sí importa.