El cuerpo necesita cobre para fabricar enzimas clave, y cuando ese mineral no se transporta bien aparecen problemas neurológicos, cutáneos y vasculares muy serios. En la enfermedad de Menkes, el problema no es la dieta, sino el modo en que el organismo distribuye el cobre; por eso cerebro, piel, pelo y tejido conectivo quedan en desventaja desde muy temprano. Aquí voy a explicar qué la causa, cómo se reconoce, qué pruebas la confirman y qué tratamiento puede cambiar el pronóstico si se inicia a tiempo.
Lo esencial para entender este trastorno del cobre y actuar pronto
- Es un trastorno genético raro del metabolismo del cobre, causado casi siempre por alteraciones en ATP7A.
- La forma clásica suele empezar en los primeros meses de vida con pelo escaso y retorcido, hipotonía, mala ganancia ponderal y convulsiones.
- El cobre sérico y la ceruloplasmina pueden orientar, pero en lactantes muy pequeños no bastan por sí solos para confirmar o descartar el cuadro.
- La confirmación más sólida la da el estudio genético, y la rapidez importa mucho porque el tratamiento precoz cambia el curso en algunos niños.
- La administración temprana de histidinato de cobre no cura la enfermedad, pero puede mejorar supervivencia y desarrollo neurológico cuando se inicia a tiempo.
- El patrón de herencia es ligado al cromosoma X, así que el consejo genético es parte real del manejo, no un trámite.
Qué falla exactamente en el transporte del cobre
Yo suelo empezar por una idea sencilla: en este trastorno no falta cobre en el mundo exterior, falta la capacidad de repartirlo bien dentro del cuerpo. El gen ATP7A codifica una proteína que mueve el cobre a través de las células; cuando esa proteína falla, el cobre se acumula donde no sirve y no llega donde sí hace falta, sobre todo al cerebro y a otros tejidos dependientes de enzimas cobre-dependientes.
Ese defecto afecta a funciones muy distintas a la vez, porque el cobre participa en la formación de vasos sanguíneos, hueso, piel, pelo y sistema nervioso. Por eso el cuadro no se limita a un solo síntoma: es una enfermedad multisistémica, con una base bioquímica clara y una expresión clínica muy desigual según la variante genética y el momento en que se detecta.
En la forma clásica, el comienzo suele ser muy temprano y la evolución puede ser rápida; en otras variantes, el problema es más lento y menos devastador. Esa diferencia es importante, porque no todos los casos pertenecen al mismo extremo del espectro. Con esa base genética en mente, ya se entiende mejor por qué los signos clínicos aparecen tan pronto y por qué el reloj diagnóstico corre tan deprisa.
Cómo suele manifestarse en los primeros meses de vida
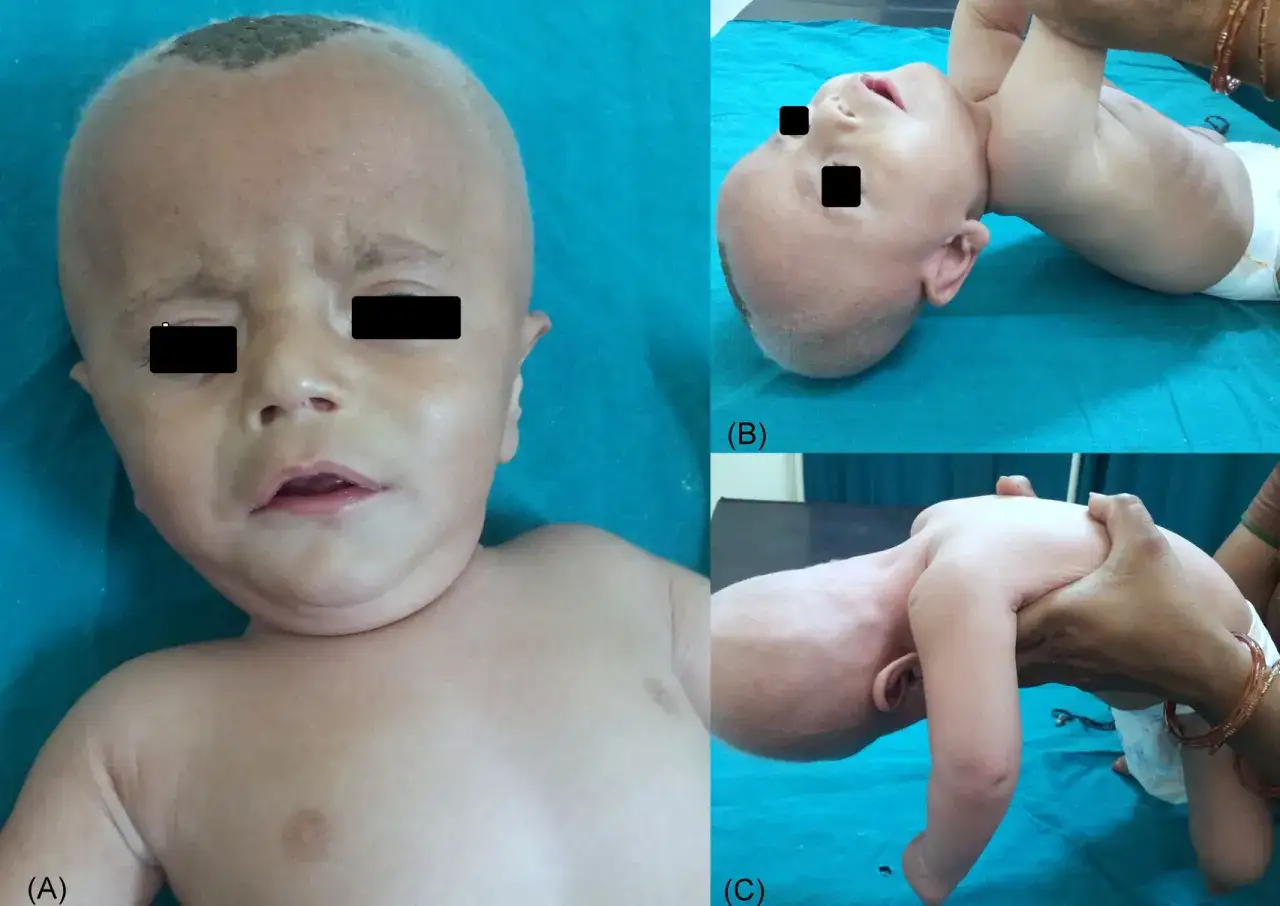
La pista más conocida es el pelo: escaso, seco, quebradizo y a menudo retorcido o ensortijado. Pero quedarse solo en el cabello sería un error. Lo que de verdad me hace sospechar es la combinación de varios hallazgos en un lactante: hipotonía, dificultad para alimentarse, falta de ganancia ponderal, irritabilidad, convulsiones y un desarrollo que empieza a ir por detrás de lo esperable.
También pueden aparecer rasgos faciales más laxos, piel y tejido conectivo alterados, temperatura corporal inestable e infecciones respiratorias repetidas. En casos más avanzados o ya floridos, el sistema vascular puede dar problemas por tortuosidad arterial, y el riesgo de hematomas subdurales o eventos cerebrovasculares no es despreciable. Cuando esos signos se juntan, la sospecha ya no es teórica.
| Forma | Inicio habitual | Rasgos dominantes | Qué implica en la práctica |
|---|---|---|---|
| Clásica | Primeros meses de vida | Pelo escaso y retorcido, hipotonía, convulsiones, retraso del desarrollo, mala ganancia de peso | Urgencia diagnóstica y terapéutica; el pronóstico depende mucho de la rapidez |
| Síndrome de cuernos occipitales | Infancia temprana o media | Piel laxa, articulaciones laxas, divertículos vesicales, tortuosidad vascular, cuernos occipitales | Curso más lento y menos grave, con peso importante de urología y rehabilitación |
| Neuropatía motora distal relacionada con ATP7A | Adolescencia o adultez | Debilidad motora distal progresiva, mínimos síntomas sensitivos | No es el cuadro clásico; el enfoque neurológico cambia bastante |
Yo me fijo mucho en la coherencia del conjunto: pelo anómalo más signos neurológicos y fallo de medro no es un detalle estético, es una señal clínica fuerte. Si esa sospecha cuaja, el siguiente paso no es esperar, sino confirmar el defecto de ATP7A con pruebas bien elegidas.
Qué pruebas confirman el diagnóstico y qué errores conviene evitar
La primera capa de estudio suele ser bioquímica: cobre sérico y ceruloplasmina. En la forma clásica, ambos suelen estar bajos, pero aquí está el error más frecuente: en menores de 8 semanas esos valores pueden ser fisiológicamente bajos incluso sin enfermedad, de modo que una interpretación apresurada puede confundir más de lo que ayuda. En otras palabras, un lactante pequeño con clínica compatible no se descarta solo porque el laboratorio todavía no sea rotundamente anormal.
La confirmación más sólida llega con el estudio genético de ATP7A. En muchos centros es la vía más directa, porque no solo confirma el diagnóstico, sino que también abre la puerta al consejo genético familiar y al diagnóstico prenatal si hace falta. Cuando hay dudas clínicas o necesidad de valorar daño neurológico, la RM cerebral y, si procede, la angio-RM pueden mostrar atrofia, mielinización retrasada y tortuosidad vascular; el EEG se reserva sobre todo para crisis epilépticas.
| Prueba | Qué aporta | Límite importante |
|---|---|---|
| Cobre sérico y ceruloplasmina | Apoyan la sospecha clínica | En los primeros 2 meses de vida pueden ser difíciles de interpretar |
| Estudio de ATP7A | Confirma la causa genética y permite estudiar a la familia | Depende de la disponibilidad del laboratorio y del tipo de variante |
| RM cerebral / angio-RM | Evalúa daño cerebral y tortuosidad vascular | No sustituye la confirmación molecular |
| EEG | Útil si hay convulsiones | Es complementario, no diagnóstico por sí solo |
Si yo tuviera que resumir esta parte en una sola idea, diría que el laboratorio ayuda, pero la genética manda. Y precisamente porque el diagnóstico no puede esperar, la pregunta siguiente siempre es la más práctica: qué tratamiento puede hacer una diferencia real.
Qué tratamiento puede cambiar el curso y cuáles son sus límites
El tratamiento específico más relevante es el histidinato de cobre administrado por vía subcutánea de forma precoz. La clave es el momento: cuando se inicia en las primeras semanas de vida, idealmente antes de que el daño neurológico se consolide, puede mejorar la supervivencia y el desarrollo neurológico en parte de los pacientes. No es una cura, pero sí una intervención que cambia el terreno de juego si llega a tiempo.
La limitación es brutalmente clara: cuando el daño ya está establecido, el cobre no lo borra. Por eso la enfermedad obliga a pensar en términos de ventana terapéutica, no de tratamiento genérico. En la forma clásica, además, el abordaje debe ser multidisciplinar y cubrir convulsiones, nutrición, disfagia, infecciones respiratorias, fisioterapia, apoyo del desarrollo y, si hace falta, gastrostomía. No se trata solo de administrar un fármaco, sino de sostener al niño desde varios frentes a la vez.
En los casos leves o variantes relacionadas con ATP7A, el enfoque cambia: la vigilancia de la vejiga, la laxitud articular, la autonomía funcional y el rendimiento escolar puede tener tanto peso como la parte neurológica. Esa diferencia explica por qué no conviene meter todos los fenotipos en el mismo saco.
Cómo se hereda y por qué el consejo genético no es un trámite
El patrón de herencia es ligado al cromosoma X. Traducido a la práctica: los varones suelen estar más afectados, las mujeres suelen ser portadoras y un padre afectado no transmite la alteración a sus hijos varones, pero sí a sus hijas. Si la madre es portadora, cada embarazo tiene un 50% de probabilidad de transmitir la variante: un hijo varón que la herede estará afectado y una hija que la herede será, en general, portadora.
También hay casos sin antecedentes familiares claros. Aproximadamente una tercera parte aparece por mutaciones nuevas, así que la ausencia de historia previa no descarta nada. Yo siempre insisto en esto porque evita dos errores muy frecuentes: culpar a la familia por “no haberlo visto venir” y subestimar el valor de estudiar a los parientes cuando ya existe una variante identificada.
Cuando la alteración familiar se conoce, pueden plantearse diagnóstico prenatal y diagnóstico genético preimplantacional. No son decisiones automáticas, pero sí opciones reales que cambian el mapa reproductivo de la familia. Y en una enfermedad tan ligada al tiempo como esta, esa información tiene un valor práctico enorme.
Con ese marco familiar claro, la última pregunta útil ya no es teórica, sino operativa: qué conviene hacer en cuanto aparece una sospecha razonable.
Qué haría yo si aparecieran signos compatibles en un lactante
Si un bebé presenta pelo anómalo, hipotonía y mala ganancia ponderal, yo lo movería como una sospecha prioritaria y no como una curiosidad dermatológica. La derivación rápida a neuropediatría y genética clínica, junto con el estudio de ATP7A, debe ir por delante de esperar a que el cuadro “se defina” solo. En esta enfermedad, esperar suele costar desarrollo neurológico.
También pediría que la familia salga de la consulta con una explicación concreta de los siguientes pasos: revisar alimentación, crisis, crecimiento, infecciones y necesidad de apoyo urológico o rehabilitador según el fenotipo. Si ya se conoce la variante familiar, el estudio de portadoras y el consejo genético deben avanzar en paralelo. Eso no solo ordena el caso actual; también evita repetir el mismo retraso en futuros embarazos o en otros familiares.
En un trastorno como este, el diagnóstico no es el final del camino, sino la pieza que permite actuar con sentido. Y cuanto antes se encaje, más opciones reales hay de modificar la historia clínica.