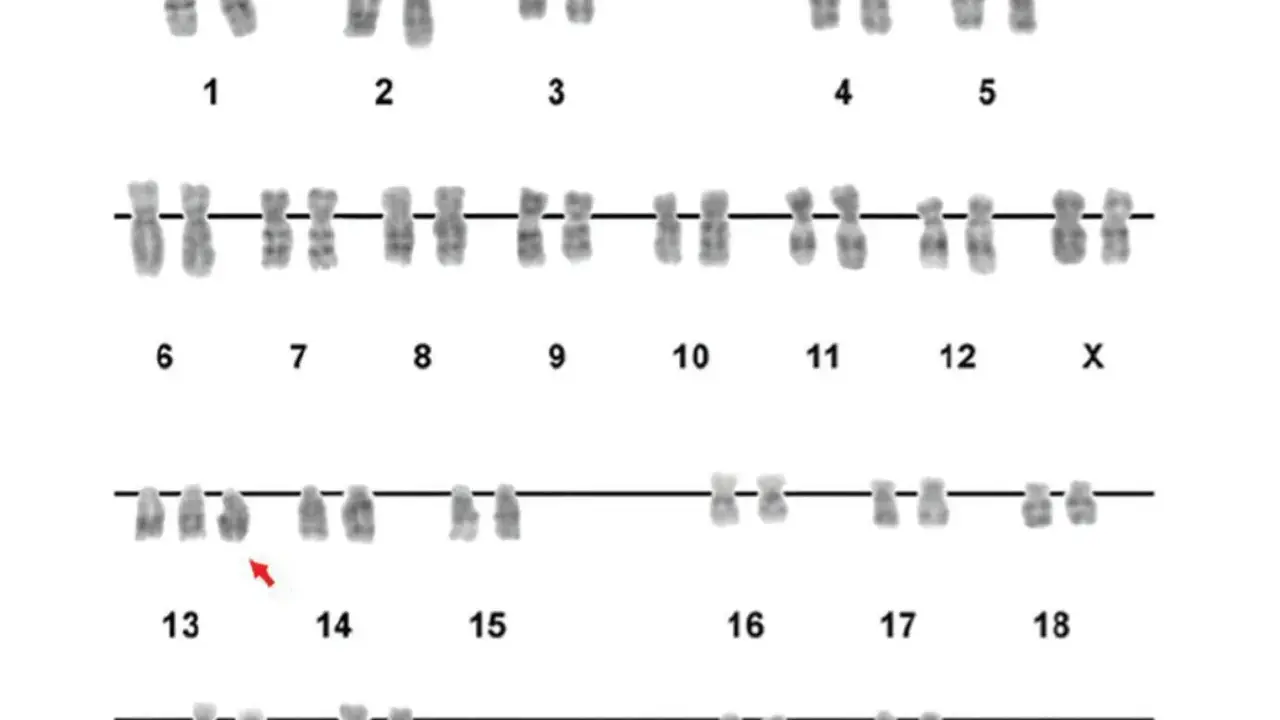
El cromosoma 13 concentra genes que intervienen en el desarrollo embrionario, la reparación del ADN y el control de la división celular. Cuando cambia su número o se pierde un fragmento, el resultado puede ir desde un síndrome cromosómico grave hasta tumores concretos o enfermedades hereditarias ligadas a genes situados en ese cromosoma. En este artículo separo esas piezas para que se entienda qué enfermedades se asocian al 13, por qué aparecen y qué pruebas se usan para estudiarlas.
Las alteraciones del cromosoma 13 no significan lo mismo en todos los pacientes
- El cromosoma 13 no es solo una cifra: su tamaño, sus genes y el tipo de alteración determinan el cuadro clínico.
- La trisomía 13 suele deberse a un error de reparto cromosómico y puede presentarse en forma completa, mosaico o translocación.
- Las deleciones del 13q se asocian a retinoblastoma, Feingold tipo 2 y otros problemas del desarrollo.
- Algunos cánceres de la sangre muestran pérdidas o translocaciones adquiridas en el cromosoma 13.
- No todo se ve en un cariotipo: a veces hace falta FISH, microarray o secuenciación de genes concretos.
Qué hace importante al cromosoma 13 en la división celular
Yo suelo explicar el cromosoma 13 con una idea sencilla: no basta con saber que existe, hay que saber si está completo, si sobra, si falta o si está roto en un punto concreto. En condiciones normales tenemos dos copias de este cromosoma, que suman entre el 3,5 % y el 4 % del ADN total de la célula y albergan unos 300 a 400 genes con funciones muy distintas. Algunos participan en el desarrollo del embrión, otros en la reparación del ADN y otros en el control fino de cómo una célula crece, se divide o se diferencia.
Cuando el error ocurre durante la meiosis, aparece una nondisjunction o no disyunción: el óvulo o el espermatozoide recibe una copia de más o de menos. Si ese gameto participa en la fecundación, el embrión puede nacer con una aneuploidía, es decir, con un número anormal de cromosomas. Si el fallo aparece más tarde, durante la mitosis de las primeras divisiones embrionarias, el resultado puede ser mosaicismo, donde unas células están alteradas y otras no. Y si lo que ocurre es un intercambio o una rotura cromosómica, hablamos de translocaciones, deleciones o duplicaciones.
- Aneuploidía: número anormal de cromosomas, como una copia extra del 13.
- Mosaicismo: coexistencia de células normales y alteradas en el mismo organismo.
- Translocación: un fragmento del cromosoma se une a otro cromosoma.
- Deleción: se pierde una parte del material genético.
Esta distinción no es académica: cambia el pronóstico, la forma de herencia y las pruebas que conviene pedir. Con esa base, la alteración más conocida es la trisomía 13, que nace precisamente de un error de reparto cromosómico y marca el inicio de varios cuadros clínicos muy distintos.
Trisomía 13 o síndrome de Patau
La trisomía 13 es la enfermedad cromosómica más clásica relacionada con este cromosoma. En la forma completa, casi todas las células tienen tres copias del 13 en lugar de dos; en la forma mosaico, solo una parte de las células tiene la copia extra; y en la forma por translocación, el material sobrante del 13 está pegado a otro cromosoma. La expresión clínica cambia mucho entre esas variantes, pero hay un patrón común: malformaciones múltiples y afectación grave del desarrollo.
| Forma | Qué ocurre | Qué suele implicar | Herencia |
|---|---|---|---|
| Trisomía completa | Hay tres copias del cromosoma 13 en casi todas las células | Suele ser la forma más grave | Por lo general aparece de forma esporádica |
| Trisomía mosaico | Solo una parte de las células tiene la copia extra | Fenotipo variable, a veces algo más leve | Habitualmente es esporádica |
| Trisomía por translocación | El material del 13 está unido a otro cromosoma | El cuadro depende de cuánto material esté duplicado | Puede heredarse si un progenitor porta una translocación equilibrada |
| Trisomía parcial | Solo una parte del cromosoma 13 está en tres copias | Síntomas variables según la región implicada | Depende del reordenamiento concreto |
Los rasgos más frecuentes incluyen cardiopatías congénitas, anomalías del cerebro o la médula espinal, microftalmia, labio leporino o paladar hendido, polidactilia y hipotonía. También son comunes las alteraciones del crecimiento y el compromiso neurológico severo. Muchos lactantes con trisomía 13 fallecen en días o semanas, y solo alrededor del 5 % al 10 % supera el primer año de vida.
La clave práctica está en no mezclar todas las formas bajo una misma etiqueta: el mosaicismo puede dar un cuadro menos uniforme, y una translocación cambia por completo la discusión genética familiar. Esa misma lógica se repite cuando el problema no es una copia extra, sino la pérdida de un tramo del 13q.
Deleciones y monosomías parciales del 13q
Una deleción no significa que falte todo el cromosoma, sino una porción concreta. Parece un detalle técnico, pero en genética clínica lo cambia todo: el tamaño del fragmento perdido, la localización exacta y los genes incluidos marcan el fenotipo. En el brazo largo del cromosoma 13 hay regiones especialmente sensibles, como 13q14 y 13q31.3, que ayudan a explicar por qué unas deleciones se asocian a retinoblastoma y otras a malformaciones de manos, pies o desarrollo neurocognitivo.
| Región afectada | Cuadro asociado | Claves clínicas | Qué conviene recordar |
|---|---|---|---|
| 13q14 | Retinoblastoma y, si la deleción es más amplia, alteraciones del desarrollo | Tumor ocular infantil, posible retraso del crecimiento, discapacidad intelectual y rasgos faciales característicos | El gen RB1 está en esta zona y su pérdida explica gran parte del riesgo tumoral |
| 13q31.3 | Síndrome de Feingold tipo 2 | Braquimesofalangia, microcefalia y dificultades de aprendizaje | La microdeleción afecta genes reguladores del desarrollo, no solo un “trozo” inespecífico |
| Deleciones o duplicaciones parciales del 13q | Monosomía o trisomía parcial | Retraso del desarrollo, bajo peso, alteraciones esqueléticas y otros rasgos físicos | La ubicación y el tamaño del cambio pesan más que el nombre genérico |
Me interesa subrayar esto porque evita errores muy comunes: dos pacientes pueden tener una “deleción del cromosoma 13” y, sin embargo, presentar problemas muy distintos. El caso de Feingold tipo 2 lo ilustra bien: no es un síndrome de afectación global y difusa, sino un patrón bastante reconocible de extremidades pequeñas, microcefalia y aprendizaje más lento. Esa precisión vuelve a ser útil cuando entramos en el terreno oncológico, donde el cambio suele ser adquirido y localizado en un clon celular.
Cambios adquiridos del cromosoma 13 en cánceres de la sangre
Aquí la lectura cambia por completo. Las alteraciones somáticas del 13 no están en todas las células del cuerpo ni se heredan como una trisomía constitucional; aparecen en células tumorales durante la vida y ayudan a explicar cómo progresa la enfermedad. La pérdida de material en 13q es frecuente en leucemias, linfomas y mieloma múltiple, y tiene valor diagnóstico o pronóstico según el contexto. En leucemia linfocítica crónica, por ejemplo, la deleción 13q14 es una de las alteraciones citogenéticas más habituales y, cuando aparece sola, suele relacionarse con un comportamiento más favorable que otras anomalías. El panorama cambia si la deleción es amplia o si coexiste con otros cambios cromosómicos.
En mieloma múltiple, la pérdida del cromosoma 13 también aparece con mucha frecuencia en el estudio citogenético, y su significado depende de si forma parte de un patrón más complejo. No es un dato decorativo: puede ayudar a estratificar riesgo y a interpretar por qué dos pacientes con el mismo diagnóstico evolucionan de forma distinta. Además, existe una alteración más específica, la translocación t(8;13), que fusiona ZMYM2 con FGFR1 y activa de manera continua vías de señalización que favorecen la proliferación celular; este cambio se ha descrito en el síndrome mieloproliferativo 8p11, que puede avanzar hacia linfoma o leucemia mieloide aguda.
- En leucemias y linfomas, la pérdida de 13q suele ser somática y limitada al clon tumoral.
- En leucemia linfocítica crónica, la deleción 13q aislada suele ser mejor que otras alteraciones, pero no todas las deleciones tienen el mismo peso.
- En mieloma múltiple, la alteración en 13q ayuda a perfilar el riesgo junto con otras anomalías.
- En el síndrome 8p11, una translocación concreta puede activar de forma constitutiva una vía oncogénica.
Lo importante aquí es entender que una alteración del cromosoma 13 puede ser hereditaria, constitucional o adquirida en el tumor. Esa diferencia cambia el estudio, el consejo familiar y el siguiente paso clínico, que es justamente lo que conviene aclarar con las pruebas adecuadas.
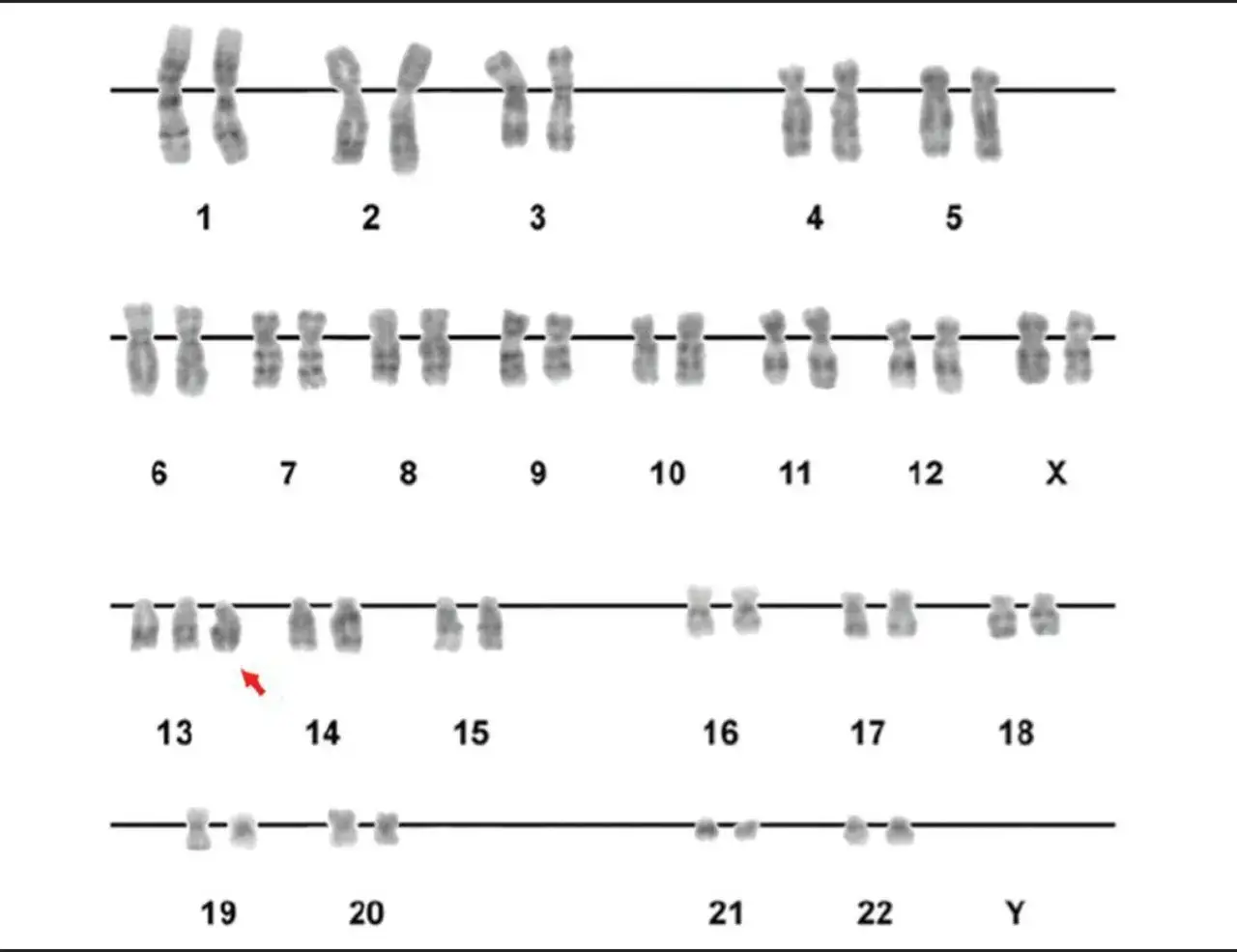
Cómo se detectan estas alteraciones en la práctica clínica
Para estudiar el cromosoma 13 no basta una sola prueba. Yo empiezo siempre por la sospecha clínica: una ecografía fetal con malformaciones, un recién nacido con rasgos compatibles con trisomía 13, un niño con retinoblastoma, un adulto con signos de Wilson o una familia con cáncer hereditario de mama y ovario son escenarios distintos y no se investigan igual. El cariotipo sigue siendo la base cuando se buscan cambios grandes en número o estructura; la hibridación in situ fluorescente, o FISH, permite mirar una región concreta con rapidez; el microarray detecta pérdidas o duplicaciones pequeñas; y la secuenciación de un gen concreto sirve cuando el problema está en la letra fina del ADN y no en el tamaño del cromosoma.
| Prueba | Qué detecta mejor | Cuándo la considero útil |
|---|---|---|
| Cariotipo | Aneuploidías, translocaciones grandes y deleciones o duplicaciones amplias | Cuando se sospecha una alteración cromosómica visible al microscopio |
| FISH | Regiones concretas o reordenamientos dirigidos | Si hay una sospecha muy específica o se necesita una respuesta rápida |
| Microarray | Microdeleciones y microduplicaciones | Cuando el cariotipo es normal pero persiste la sospecha de un cambio pequeño |
| Secuenciación de genes o paneles | Variantes en genes como RB1, ATP7B o BRCA2 | Si el problema está en un gen del cromosoma 13 y no en la estructura cromosómica |
En embarazo, un test de cribado puede avisar, pero no confirma. Cuando la sospecha es real, la confirmación se hace con vellosidades coriónicas o amniocentesis y estudio de las células fetales. Y aquí hay una idea que conviene no olvidar: un cariotipo normal no descarta una enfermedad causada por un gen del cromosoma 13, como Wilson o los síndromes asociados a BRCA2. Por eso la elección de la prueba no debería ser automática, sino guiada por el patrón clínico.
Una vez identificado el tipo de alteración, la pregunta útil ya no es solo qué prueba salió positiva, sino qué cambia en el manejo, el seguimiento y el riesgo familiar. Ahí es donde el consejo genético deja de ser un complemento y pasa a ser parte del diagnóstico.
Qué cambia para el tratamiento y el consejo genético
El tratamiento depende menos del cromosoma en abstracto que del cambio exacto y del órgano afectado. En la trisomía 13 no existe una cura específica; el manejo se centra en el soporte clínico y en decisiones muy individualizadas. En retinoblastoma, detectar una deleción de 13q a tiempo puede cambiar el pronóstico ocular y orientar el estudio familiar. En Wilson, identificar mutaciones en ATP7B permite iniciar un tratamiento que evita la acumulación de cobre en hígado, cerebro y ojos. Y en BRCA2, el hallazgo no sirve para tratar un tumor ya presente, pero sí para intensificar la vigilancia, valorar prevención y estudiar a los familiares que puedan compartir el riesgo.
La distinción entre alteración constitucional y alteración somática es clave. Si el cambio está en todas o casi todas las células, puede influir en embarazos futuros y justificar un estudio de los progenitores. Si la alteración es somática y aparece solo en el tumor, el impacto hereditario suele ser mucho menor. Y si lo que hay es una variante en un gen concreto del cromosoma 13, la interpretación ya no gira en torno al cromosoma completo, sino al órgano diana y al patrón de herencia de ese gen.
- Alteración constitucional: puede afectar el desarrollo y el riesgo reproductivo.
- Alteración somática: orienta el pronóstico oncohematológico y no suele pasar a la descendencia.
- Variante génica: exige prevención o seguimiento específico según el gen afectado.
Si hay un progenitor portador de una translocación equilibrada, el riesgo de recurrencia puede aumentar de forma relevante, aunque el portador esté sano. Si la alteración es de novo, ese riesgo suele bajar, pero no desaparece por completo hasta que se interpreta el caso con detalle. Por eso insisto tanto en no etiquetar a la ligera cualquier hallazgo del cromosoma 13 como “la misma enfermedad”.
Lo que más conviene retener cuando el cromosoma 13 sale alterado
La primera idea es que no existe una sola “enfermedad del cromosoma 13”, sino varios escenarios distintos: trisomía completa, mosaicismo, translocaciones, deleciones parciales, cambios adquiridos en tumores y variantes en genes concretos. La segunda es que la localización importa tanto como el tipo de cambio; perder 13q14 no significa lo mismo que perder 13q31.3, y una mutación en RB1 no se interpreta igual que una en ATP7B o BRCA2. La tercera es que el dato más valioso rara vez es el nombre de la alteración, sino su contexto biológico y clínico.
Si yo tuviera que resumirlo en una sola frase, diría que el cromosoma 13 se entiende mejor cuando se lee como un mapa y no como una etiqueta. Número de copias, región afectada, tipo de célula, momento en que aparece el cambio y edad del paciente: esos cinco datos suelen pesar más que cualquier titular genérico. Y es justamente ahí donde la genética clínica aporta más valor, porque transforma un hallazgo cromosómico en decisiones reales para el paciente y para su familia.