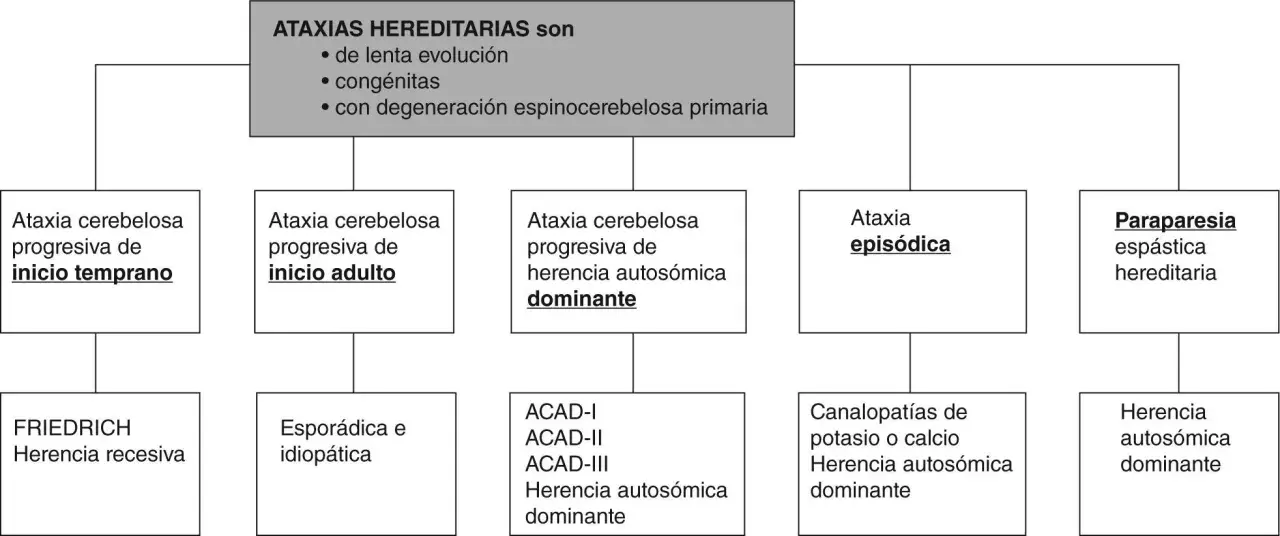
Lo esencial sobre la herencia de la ataxia de Friedreich
- Patrón clave: es una enfermedad de herencia autosómica recesiva.
- Gen implicado: la causa está en FXN, que codifica la frataxina.
- Forma más habitual: una expansión de repeticiones GAA en el intrón 1 del gen.
- Riesgo por embarazo: si ambos progenitores son portadores, hay un 25% de riesgo de un hijo afectado, un 50% de portador y un 25% de no portador.
- Portadores: suelen estar asintomáticos y no desarrollan la enfermedad.
- Prueba adecuada: no basta con una secuenciación estándar; hay que estudiar la expansión GAA y, si hace falta, variantes o deleciones de FXN.
Cómo se transmite de padres a hijos
Cuando explico esta enfermedad, empiezo por lo más importante: es autosómica recesiva. Eso significa que el gen implicado, FXN, no está en los cromosomas sexuales, así que hombres y mujeres tienen el mismo riesgo. Para que aparezca la enfermedad, la persona debe heredar dos copias alteradas, una de cada progenitor. Si solo recibe una, suele ser portadora y no desarrolla el cuadro.
Ese detalle aclara por qué a veces la historia familiar parece “saltar” una generación: dos padres sin síntomas pueden transmitir, sin saberlo, una variante patogénica cada uno. En poblaciones de ascendencia europea, la frecuencia de portadores se ha estimado aproximadamente entre 1/60 y 1/100, así que no hace falta un historial llamativo para que aparezca un caso en la familia. También conviene desmontar otra idea frecuente: aunque la frataxina participa en la función mitocondrial, no se trata de una herencia mitocondrial. El error está en el núcleo, en un gen autosómico, no en el ADN de las mitocondrias. Y justo ahí aparece la siguiente pregunta: qué tipo de alteración concreta hay en FXN.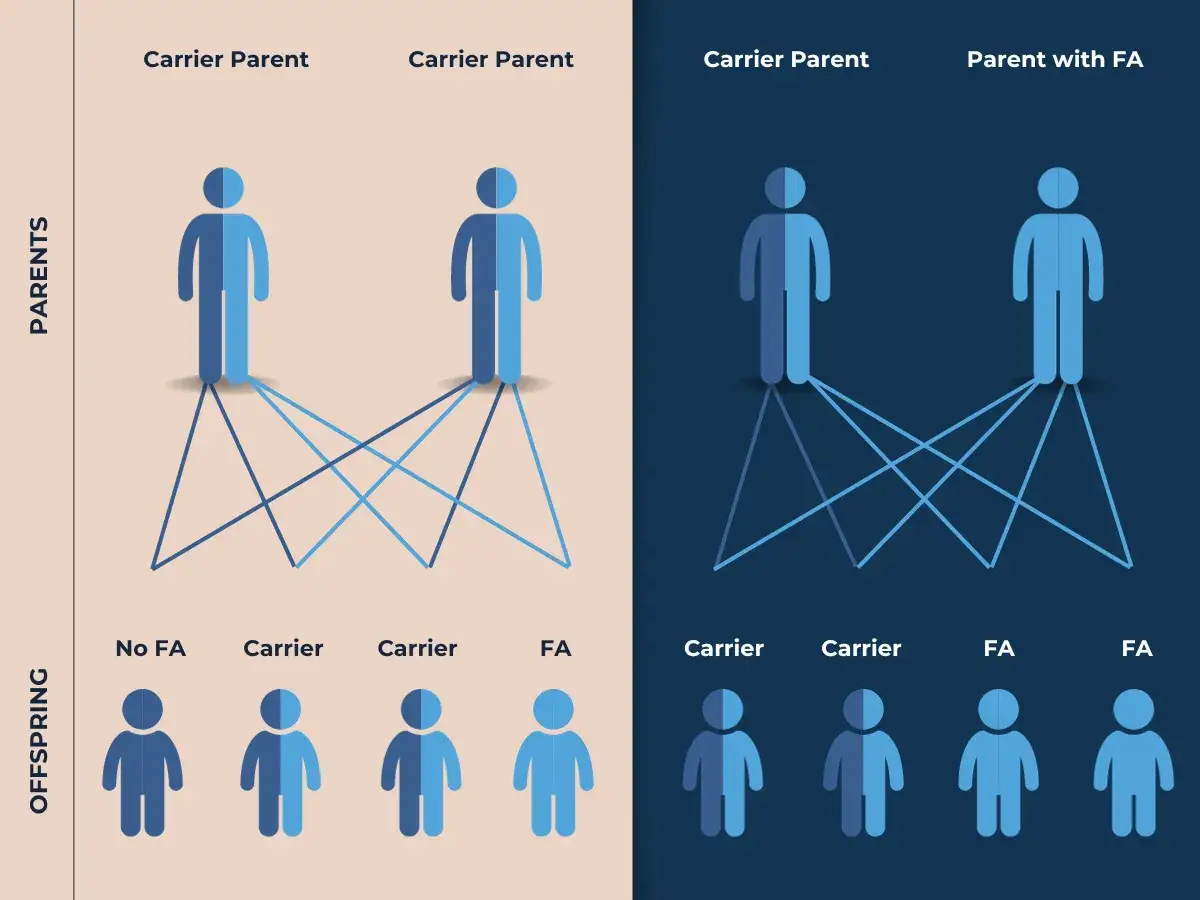
Qué cambia en el gen FXN
En la práctica, la alteración más habitual no es una sustitución aislada, sino una expansión de tripletes GAA en el intrón 1 del gen FXN. En personas sin la enfermedad, ese tramo suele ser corto; en los alelos patogénicos puede llegar a cientos o incluso superar el millar de repeticiones. La consecuencia es una producción insuficiente de frataxina, una proteína necesaria para el buen funcionamiento celular.
La relación no es puramente binaria: cuanto más larga es la expansión, más probable es un inicio más temprano y una evolución más intensa, aunque el tamaño del alelo no lo explica todo por sí solo. De forma orientativa, los alelos más cortos dentro del rango patogénico tienden a asociarse con un debut más tardío que las expansiones largas. Eso ayuda a entender por qué hay formas de inicio infantil, adolescente o incluso adulto, pero no cambia el patrón de herencia de base.
| Tipo de alteración | Frecuencia aproximada | Lectura práctica |
|---|---|---|
| Expansión GAA en ambas copias | ~96% | Forma clásica de la enfermedad |
| Expansión GAA + variante patogénica o deleción | ~4% | Compuesto heterocigoto, con expresión variable |
| Dos variantes puntuales sin expansión | Excepcional | Situación muy rara, descrita en familias concretas |
Qué riesgo real tiene cada embarazo
La regla mendeliana aquí es sencilla, pero suele confundirse en la consulta. Si ambos progenitores son portadores, cada embarazo tiene un 25% de probabilidad de dar un hijo afectado, un 50% de dar un portador sano y un 25% de no heredar ninguna de las variantes familiares. Lo importante es que ese cálculo se repite en cada gestación: un hijo sano no “gasta” la probabilidad del siguiente.
| Situación familiar | Probabilidad de hijo afectado | Qué suele ocurrir |
|---|---|---|
| Ambos progenitores son portadores | 25% | Cada embarazo tiene un 25% de riesgo, un 50% de portador y un 25% de no portador |
| Un progenitor es portador y el otro no lo es | 0% | No habrá hijos afectados, pero sí un 50% de portadores |
| Una persona afectada y su pareja no es portadora | 0% | Todos los hijos serán portadores, pero no enfermos |
| Una persona afectada y su pareja es portadora | 50% | La mitad de los hijos puede estar afectada y la otra mitad será portadora |
En familias reales, la pregunta no siempre es “¿lo tendré o no?”, sino “¿debo estudiar a mi pareja, a mis hermanos o a mis hijos adultos?”. Mi criterio es directo: si existe una variante familiar conocida, primero se aclara esa variante y después se decide a quién estudiar. Así se evita una cascada de resultados ambiguos y se gana precisión. Eso nos lleva a la parte más subestimada: cómo interpretar bien la prueba genética para no sobrerreaccionar ni infravalorar un resultado.
Cómo se interpreta el estudio genético
Para esta enfermedad, la palabra clave es método. El estudio adecuado suele empezar por la expansión GAA en FXN y, si hace falta, se completa con secuenciación y análisis de deleciones o duplicaciones. Un exoma sin más no es suficiente para descartar la causa clásica, porque la expansión intrónica no se detecta de forma fiable con esa técnica.| Prueba | Qué detecta | Cuándo conviene |
|---|---|---|
| Análisis específico de repetición GAA | Número de repeticiones en FXN | Primera elección ante sospecha clínica o antecedente familiar |
| Secuenciación de FXN | Variantes puntuales y pequeñas inserciones o deleciones | Cuando una expansión sola no explica el cuadro o hay que completar el estudio |
| Análisis de deleción/duplicación | Grandes deleciones o cambios de número de copias | Si la sospecha persiste y la secuenciación no resuelve el caso |
| Panel multigénico de ataxias | Varios genes relacionados con ataxia hereditaria | Cuando el fenotipo no encaja de forma clara con ataxia de Friedreich |
Ojo con el exoma: puede ser útil para otras ataxias, pero no suele resolver por sí solo la expansión GAA intrónica. Si el laboratorio no informa específicamente sobre repeticiones, un resultado normal no cierra la pregunta. Y si ya se conoce la alteración familiar, el estudio dirigido a esa variante es mucho más limpio para valorar portadores. Esta diferencia parece obvia, pero en la práctica evita mucha ansiedad innecesaria.
Los malentendidos que más confunden a las familias
Yo vigilaría especialmente estos errores, porque son los que más distorsionan las decisiones familiares:
- “Portador” no es “enfermo”. Una sola copia alterada suele no dar síntomas, pero sí puede pasar a la descendencia.
- Que los padres estén sanos no cambia el riesgo. Precisamente porque el patrón es recesivo, los progenitores suelen ser portadores asintomáticos.
- Un exoma normal no descarta la enfermedad. La expansión GAA necesita un análisis específico.
- No es una herencia ligada al sexo ni mitocondrial. El gen está en un autosoma, así que el sexo no modifica la probabilidad de transmitirlo.
- El tamaño de la expansión importa, pero no lo explica todo. Ayuda a orientar el inicio y la severidad, pero no sustituye la valoración clínica.
- Los alelos intermedios exigen interpretación experta. No todo resultado fuera de lo habitual equivale automáticamente a enfermedad.
Cuando esa lectura se hace bien, la conversación con la familia deja de girar en torno al miedo y pasa a decisiones concretas. Y eso es justo lo que conviene ordenar antes de cerrar un plan reproductivo.
Lo que yo revisaría antes de cerrar un plan familiar
Si hay antecedentes, yo ordenaría el proceso así:
- Confirmar el diagnóstico del caso índice y conservar el informe genético original.
- Identificar la alteración exacta en FXN para poder hacer un estudio dirigido.
- Estudiar a la pareja si el objetivo es estimar riesgo reproductivo real.
- Valorar asesoramiento genético para traducir el resultado en números comprensibles y en escenarios de riesgo.
- Explorar, si la familia lo desea, opciones como diagnóstico prenatal o diagnóstico genético preimplantacional, siempre con una conversación clara sobre límites, tiempos y costes emocionales.
En España, como en cualquier contexto clínico serio, el valor del consejo genético está en convertir un informe técnico en decisiones comprensibles. Si hay un caso en la familia, yo no empezaría por suposiciones, sino por la variante concreta y por un cálculo de riesgo bien hecho; eso cambia por completo la calidad de la conversación y evita interpretaciones erróneas que luego son difíciles de corregir.