La lectura de un gel de agarosa no consiste solo en ver bandas sobre un fondo claro: consiste en decidir si un fragmento de ADN tiene el tamaño esperado, si la muestra está íntegra y si el ensayo genético merece pasar al siguiente paso. En PCR, digestiones de restricción o verificación de plásmidos, una mala interpretación puede hacerte descartar una muestra válida o aceptar una señal engañosa. Aquí te explico cómo leo yo un gel, qué indica cada patrón y dónde suelen producirse los errores.
Lo que debes tener claro antes de fijarte en las bandas
- La posición de la banda te habla del tamaño relativo, no de una variante concreta.
- La intensidad sugiere cantidad de ADN, pero no sirve para cuantificar con precisión sin una referencia.
- El marcador de peso molecular es tu escala; sin él, la lectura pierde mucha fiabilidad.
- Un smear, una banda débil o una banda extra no significan lo mismo en una PCR, un digest o un plásmido sin cortar.
- Si la prueba tiene implicaciones clínicas, el gel suele ser una verificación previa, no la última palabra.
Qué te dice realmente un gel de agarosa en genética
La electroforesis en gel de agarosa separa ácidos nucleicos por tamaño: el ADN migra hacia el polo positivo y los fragmentos pequeños avanzan más rápido que los grandes. Eso parece simple, pero en genética importa mucho porque el gel no “lee” una mutación por sí mismo; lo que muestra es si un fragmento está donde debería estar, si la amplificación ha funcionado y si la muestra está lo bastante limpia e íntegra.
Yo lo trato como una prueba de coherencia. Si una PCR estaba diseñada para generar un amplicón de 420 pb y en el carril aparece una banda nítida en ese rango, la reacción probablemente ha ido bien. Si aparece una banda en otro tamaño, varias bandas o una señal difusa, ya no estoy ante un resultado limpio, sino ante una pista que tengo que contrastar con el diseño del ensayo y con los controles.
Hay una diferencia importante entre “se ve ADN” y “el resultado es interpretable”. En un contexto de pruebas genéticas, un gel puede servir para confirmar una amplificación, revisar una digestión de restricción, comprobar la integridad del ADN genómico o verificar un plásmido; lo que no hace es sustituir a la secuenciación, a una prueba específica de variante o a la validación clínica cuando el hallazgo tiene peso médico.
Con esa base, ya se puede pasar a la lectura real de las bandas sin caer en conclusiones demasiado rápidas.
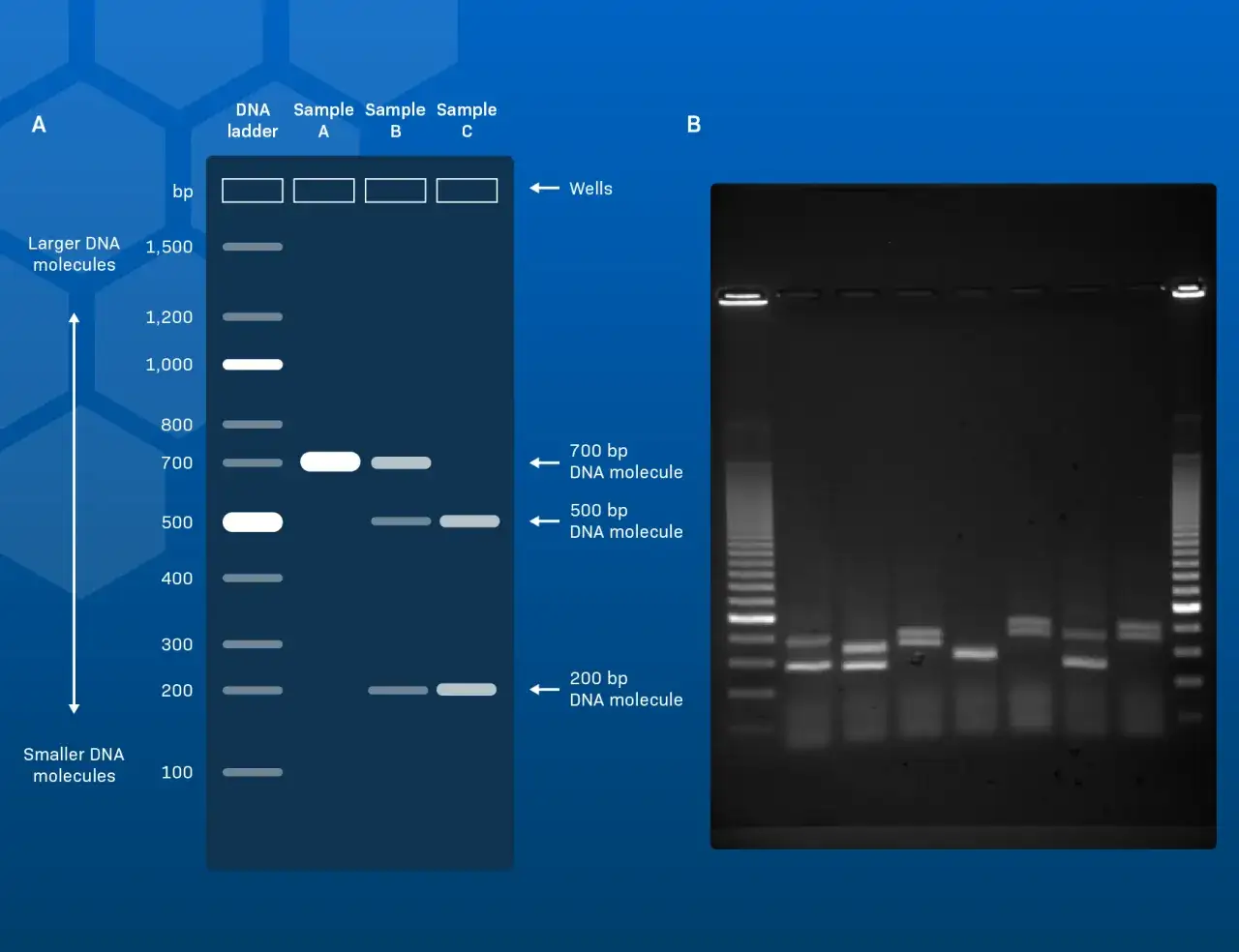
Cómo leo una banda paso a paso
Yo empiezo siempre por el carril del marcador, porque sin él cualquier estimación de tamaño es una intuición. Después miro la posición de la banda respecto a ese patrón, comparo la intensidad con los controles y, por último, reviso la forma: una banda compacta, una banda difusa o un arrastre largo no cuentan la misma historia.
| Qué miro | Qué significa | Qué compruebo después |
|---|---|---|
| Marcador de peso molecular | Me da la escala de tamaños para comparar | Si es el ladder adecuado para el rango esperado |
| Posición de la banda | Me orienta sobre el tamaño aproximado del fragmento | Si coincide con el amplicón o fragmento esperado |
| Intensidad | Suele reflejar más o menos ADN, pero no cuantificación exacta | Si hubo sobrecarga, poca muestra o tinción desigual |
| Anchura y nitidez | Una banda estrecha suele ser más interpretable | Si hay degradación, sales, exceso de ADN o corrido deficiente |
| Controles | Indican si el ensayo es válido | Si el positivo amplifica y el negativo sigue limpio |
Como guía práctica, suelo elegir el gel según el tamaño del fragmento que quiero separar: 0,7-0,8% para fragmentos grandes, 1% para rangos intermedios y 1,5-2% cuando me interesa resolver fragmentos pequeños y muy próximos entre sí. En la misma línea, si las bandas están demasiado juntas, me resulta más útil cambiar el porcentaje del gel o dejar correr más tiempo que subir sin más el voltaje.
Ese paso de “mirar dónde está la banda” es solo el comienzo; cuando ya sabes leer posición y tamaño, toca separar patrones útiles de artefactos.
Patrones frecuentes y qué suelen indicar
La parte más útil, y la que más errores evita, es aprender a reconocer patrones. En genética, una imagen bonita no siempre es una imagen correcta: a veces una señal débil es aceptable, y a veces una banda muy intensa es justamente el problema.
| Patrón | Qué suele indicar | Cómo lo interpreto yo |
|---|---|---|
| Banda única y nítida en el tamaño esperado | Amplificación o digestión coherente | Es el escenario más limpio, aunque sigo mirando controles y diseño experimental |
| Varias bandas | Amplificación inespecífica, digestión incompleta o mezcla de moléculas | No doy por válido el resultado hasta saber cuál es la banda correcta |
| Smear o arrastre | Degradación, sobrecarga, sales, ADN mal purificado o corrido agresivo | Me dice que la muestra o la corrida no están lo bastante limpias |
| Banda muy débil o ausente | Poca cantidad de ADN, reacción fallida o problema de tinción/carga | No la leo como negativo automático si falta control interno |
| Bandas curvadas o “sonrisa” | Sobrecalentamiento o distribución desigual del campo eléctrico | Sospecho del corrido antes que de la muestra |
| Marcador borroso o extraño | Problema del ladder, del buffer o de la preparación del gel | Si el control falla, la muestra pierde valor interpretativo |
En la práctica, la señal más engañosa es la ausencia de banda. En una PCR diagnóstica, por ejemplo, una muestra sin amplicón no es concluyente si tampoco funciona el control interno; puede significar inhibición, ADN insuficiente o un error técnico. Y en un gel de plásmidos, una banda más arriba o más abajo de lo previsto no siempre es “un error de tamaño”: a veces es solo una conformación diferente de la molécula.
Ese mapa de errores cambia un poco según el tipo de prueba genética, y ahí es donde mucha gente se precipita.
Qué cambia según la prueba genética
No interpreto igual una PCR, una digestión de restricción o un ADN plasmídico sin cortar. En genética clínica y molecular, el contexto del ensayo manda: el mismo patrón puede ser correcto en un caso y problemático en otro.
| Tipo de prueba | Qué espero ver | Qué me hace sospechar |
|---|---|---|
| PCR convencional | Una o pocas bandas en el tamaño previsto | Bandas extra, smear o ausencia de banda con controles válidos |
| Digestión de restricción | Fragmentos que coinciden con el mapa esperado | Bandas faltantes, fragmentos del tamaño incorrecto o digestión parcial |
| Plásmido sin cortar | Varias conformaciones posibles: superenrollada, relajada y lineal | Si lo leo como un fragmento lineal simple, me puedo equivocar |
| ADN genómico | Material de alto peso molecular cerca del pocillo | Un smear amplio sugiere degradación o cizallamiento |
| Ensayos de presencia/ausencia | Una banda diana y un control interno | Sin control interno, la ausencia de banda no demuestra nada por sí sola |
Hay un límite técnico que conviene tener muy presente: el gel resuelve tamaño y, en parte, calidad, pero no identifica una variante puntual con precisión clínica. Si el objetivo es detectar una mutación concreta, un cambio de una sola base o una alteración pequeña, yo no confiaría solo en la agarosa; pediría secuenciación, una sonda específica, qPCR o el método ortogonal que corresponda.
Con ese contexto, los fallos de lectura más comunes se detectan antes y se corrigen mejor.
Errores de interpretación que veo con más frecuencia
Cuando reviso resultados de gel en pruebas genéticas, hay varios tropiezos que se repiten mucho. No suelen deberse a falta de experiencia en una sola cosa, sino a querer leer más de lo que la imagen realmente ofrece.
- Confundir intensidad con cantidad exacta. Una banda más brillante suele sugerir más ADN, pero la tinción, la saturación y la carga también influyen.
- Tomar un negativo como definitivo sin control interno. Si el control falla, la ausencia de banda no demuestra ausencia de la secuencia diana.
- Elegir un marcador que no cubre el rango esperado. Si el ladder no está pensado para ese tamaño, la estimación se vuelve floja desde el principio.
- Leer un plásmido sin cortar como si fuera ADN lineal. La conformación modifica la migración y puede hacer que el tamaño aparente engañe.
- Cargar demasiado ADN. Las bandas se ensanchan, aparecen arrastres y la resolución cae justo donde más la necesitas.
- Ignorar el aspecto del gel entero. Si el marcador está borroso o la banda del centro “sonríe”, el problema puede estar en el corrido, no en la muestra.
- Dar por buena una banda extra sin buscar la causa. En una PCR, una señal adicional puede ser inespecificidad; en un digest, puede ser corte incompleto.
Yo suelo resumirlo así: si la imagen no encaja con el diseño experimental, primero sospecho del sistema, después de la muestra y solo al final de la conclusión. Ese orden ahorra repeticiones innecesarias y evita decisiones demasiado rápidas.
Si después de estas comprobaciones la imagen sigue sin encajar, yo no la daría por concluyente.
La revisión que haría antes de dar un resultado por bueno
Antes de aceptar un gel como válido, sigo una revisión corta pero estricta. No hace falta complicarla: basta con comprobar que cada pieza sostiene a la siguiente.
- ¿El marcador de peso molecular cubre el rango que necesito interpretar?
- ¿El control positivo muestra la banda esperada?
- ¿El control negativo está limpio o aparece contaminación?
- ¿La banda diana coincide con el tamaño previsto por el diseño del ensayo?
- ¿La forma de la banda es nítida o hay smear, sobrecarga o “smiling”?
- ¿La interpretación exige confirmación adicional con secuenciación, qPCR u otro método?
Si una banda importante me deja dudas, yo la trato como una hipótesis técnica, no como un resultado cerrado. Esa disciplina evita errores de diagnóstico, repeticiones innecesarias y lecturas demasiado optimistas de un gel que, por diseño, solo responde bien cuando la muestra, el control y el contexto están en armonía.