La mucopolisacaridosis tipo III, más conocida como síndrome de Sanfilippo, es una enfermedad genética rara que afecta sobre todo al cerebro y suele manifestarse en la infancia con retraso del lenguaje, cambios de conducta y pérdida progresiva de habilidades ya adquiridas. Aquí explico qué la causa, cómo reconocer sus señales tempranas, cómo se diagnostica y qué se puede hacer hoy para acompañar mejor al niño y a su familia.
Lo esencial para entender una enfermedad neurodegenerativa rara que empieza en la infancia
- Afecta sobre todo al sistema nervioso central y puede pasar desapercibida al nacer.
- La señal más útil suele ser la regresión: pérdida de lenguaje, aprendizaje o conducta previamente adquiridos.
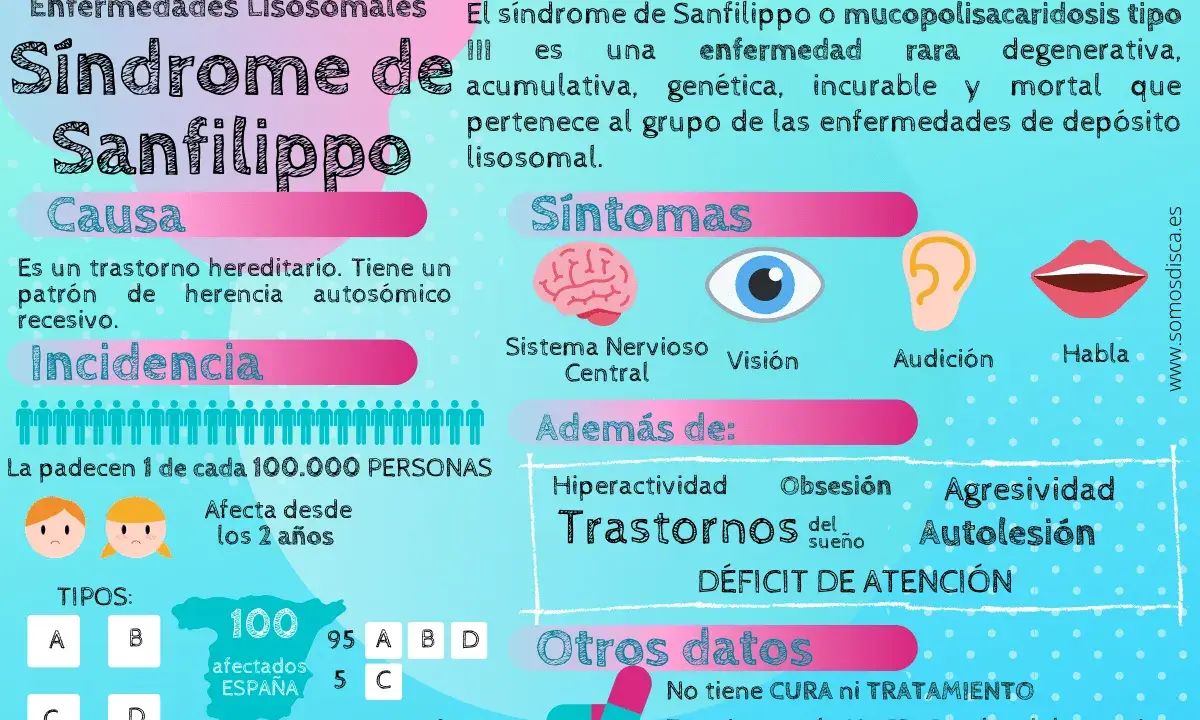
- Su base es genética, con herencia autosómica recesiva y cuatro subtipos clásicos: A, B, C y D.
- El diagnóstico combina sospecha clínica, análisis de orina, pruebas enzimáticas y estudio genético.
- Hoy no existe una cura aprobada que frene la enfermedad, pero sí cuidados de apoyo que cambian mucho la calidad de vida.
Qué es la mucopolisacaridosis tipo III y por qué cambia tanto la vida familiar
Yo suelo explicarla así: el problema no está en cómo nace el niño, sino en cómo sus células reciclan ciertas moléculas. En esta enfermedad faltan o funcionan mal unas enzimas lisosomales que degradan el heparán sulfato, un tipo de glucosaminoglicano; cuando ese material se acumula dentro de las células, daña sobre todo al cerebro y al sistema nervioso central. Por eso hablamos de un trastorno de almacenamiento lisosomal, no de un simple retraso del desarrollo.
Lo más desconcertante para muchas familias es que el niño puede parecer normal al principio. No suele haber un aspecto llamativo desde el nacimiento, y los cambios importantes aparecen más tarde, cuando ya se esperaba una evolución normal del habla, del sueño o de la conducta. Esa mezcla de inicio silencioso y progresión neurológica es precisamente lo que hace que el diagnóstico se retrase con facilidad.Además, aunque el nombre técnico sea largo, la idea clínica es bastante concreta: la enfermedad afecta sobre todo al cerebro, pero no solo al cerebro. También pueden aparecer diarrea crónica, infecciones repetidas, hernias, rigidez articular o problemas auditivos y visuales. Esa combinación explica por qué la valoración debe ser integral y no centrarse solo en una consulta aislada. Y precisamente por eso las primeras señales suelen parecer “otra cosa”, no una enfermedad genética rara.
Las señales tempranas que suelen pasar desapercibidas
Si tuviera que señalar una pista que cambia el rumbo clínico, sería esta: el niño no solo va más despacio, sino que pierde habilidades que ya tenía. No es lo mismo un retraso aislado que una regresión. Esa diferencia, que a veces parece sutil, es la que debe hacer saltar las alarmas.
- Retraso del lenguaje que no mejora como se esperaba.
- Pérdida de palabras, gestos o habilidades sociales ya adquiridas.
- Sueño fragmentado, despertares frecuentes o insomnio persistente.
- Hiperactividad, impulsividad, conductas agresivas o muy temerarias.
- Rasgos que pueden parecer del espectro autista, sobre todo si hay dificultades de comunicación.
- Diarrea crónica, otitis o infecciones de garganta repetidas.
- Torpeza motora, rigidez, hernias, pérdida auditiva o problemas de visión.
El error más común es explicar cada signo por separado: “ya hablará”, “duerme mal por la edad”, “es muy movido”, “se porta mal”. Yo no me quedaría ahí si varios de esos elementos aparecen juntos o si hay una regresión clara. En ese escenario, la causa orgánica debe entrar pronto en la conversación clínica, porque el tiempo perdido rara vez vuelve. Cuando ese patrón aparece, la siguiente pieza del puzzle es genética.
La base genética y las diferencias entre los tipos A, B, C y D
La enfermedad se hereda con un patrón autosómico recesivo. Eso significa que, en la mayoría de los casos, ambos progenitores son portadores de una copia alterada del gen sin presentar síntomas. En cada embarazo, la probabilidad es de 25% de tener un hijo afectado, 50% de que sea portador y 25% de que no herede la variante familiar.
La consanguinidad no causa la enfermedad por sí sola, pero aumenta la probabilidad de que dos personas compartan la misma variante genética rara. Cuando existe antecedente familiar, el consejo genético deja de ser un accesorio y pasa a ser parte central de la atención.
| Tipo | Gen afectado | Enzima implicada | Tendencia clínica |
|---|---|---|---|
| A | SGSH | Heparán N-sulfatasa | Suele empezar antes y progresar con más rapidez. |
| B | NAGLU | Alfa-N-acetilglucosaminidasa | Curso variable; puede ser severo. |
| C | HGSNAT | Heparán acetil-CoA:alfa-glucosaminida N-acetiltransferasa | También es progresivo, con un ritmo que puede ser algo más lento en algunos casos. |
| D | GNS | N-acetilglucosamina-6-sulfatasa | Es el subtipo más infrecuente dentro del grupo clásico. |
La clave práctica es que los síntomas se parecen mucho entre sí, pero el ritmo de progresión no. En series clínicas españolas, el tipo A suele aparecer con más frecuencia y tiende a ser el más agresivo. Esa diferencia no cambia solo el pronóstico; también orienta qué esperar, qué vigilar y cómo hablar con la familia sin prometer más de lo que la evidencia permite. Saber el subtipo ayuda, pero no sustituye la confirmación diagnóstica.
Cómo se diagnostica en la práctica clínica
El diagnóstico no debería apoyarse en una única prueba. Yo prefiero pensar en una secuencia: primero la sospecha clínica, después la confirmación bioquímica y, por último, la confirmación genética. Si una familia llega a consulta con retraso del habla, conducta difícil y regresión, no conviene esperar a que la imagen clínica “se complete”.
- Sospecha clínica: retraso del lenguaje, regresión, alteraciones del sueño, conducta atípica y otros signos sistémicos.
- Análisis de orina: suele buscar glucosaminoglicanos, especialmente heparán sulfato, como pista inicial.
- Estudio enzimático: mide la actividad de la enzima sospechosa en sangre, leucocitos o fibroblastos.
- Confirmación molecular: identifica la variante en SGSH, NAGLU, HGSNAT o GNS y define el subtipo.
- Valoración complementaria: audición, visión, corazón, estado neurológico y, si hace falta, neuroimagen.
La orina orienta, pero no cierra el caso. Si yo tuviera que resumir una regla práctica, sería esta: cuando la sospecha es alta, hay que estudiar en paralelo, no ir paso a paso durante meses. Eso reduce retrasos innecesarios y evita que el niño llegue tarde a una atención especializada. Y una vez puesto el nombre a la enfermedad, la pregunta pasa de “qué es” a “qué podemos hacer hoy”.
Qué se puede hacer hoy para tratarlo y acompañar mejor al niño
Hoy el enfoque es sintomático y multidisciplinar. No existe todavía un tratamiento curativo aprobado que detenga de forma fiable la neurodegeneración, así que el trabajo real se centra en aliviar síntomas, preservar funciones el mayor tiempo posible y sostener a la familia. Esa combinación, aunque menos espectacular que una promesa terapéutica, es la que de verdad cambia el día a día.
- Sueño: rutinas estables, entorno predecible y tratamiento cuando el especialista lo indica.
- Comunicación: logopedia y sistemas aumentativos o alternativos de comunicación cuando el lenguaje oral empieza a fallar.
- Conducta: estrategias estructuradas, evitar castigos inútiles y adaptar expectativas al perfil neurológico real del niño.
- Motricidad: fisioterapia y terapia ocupacional para mantener movilidad, equilibrio y autonomía el máximo tiempo posible.
- Audición y visión: revisiones periódicas, porque perder visión o audición empeora mucho la interacción y la conducta.
- Nutrición y deglución: valorar textura de alimentos, riesgo de atragantamiento y apoyo nutricional si aparece disfagia.
- Crisis epilépticas y dolor: seguimiento neurológico y ajuste de medicación cuando sea necesario.
- Apoyo familiar: trabajo social, respiro familiar, adaptación escolar y, cuando corresponde, cuidados paliativos tempranos.
Quiero subrayar algo importante: cuidados paliativos no significa rendirse. En una enfermedad progresiva, anticiparse al dolor, al insomnio, a la disfagia y al agotamiento del cuidador forma parte de una buena medicina. También es útil revisar qué ofrece la escuela, qué apoyos cubre el sistema sanitario y qué grado de dependencia o discapacidad se puede reconocer, porque eso cambia el acceso a recursos reales.
Y sí, la tentación de buscar una única solución es enorme. Pero, a día de hoy, lo que más impacto tiene no es un fármaco aislado, sino la coordinación entre pediatría, neurología, genética, rehabilitación y apoyo psicosocial. Esa coordinación prepara el terreno para entender mejor la investigación que está llegando.
Qué está cambiando en 2026 y qué pronóstico conviene esperar
En 2026, el mensaje honesto es claro: hay investigación en marcha, pero todavía no una cura aprobada. Siguen activos estudios sobre terapia génica, reemplazo enzimático, terapias celulares y estrategias dirigidas a síntomas neuroconductuales, como algunos ensayos con cannabidiol. Eso no significa que ya exista una solución disponible en la práctica habitual; significa que el campo se está moviendo, pero con criterios de elegibilidad estrictos y resultados que todavía hay que consolidar.
Conviene ser prudente con las expectativas. Un ensayo clínico no equivale a tratamiento estándar, y una terapia prometedora en fase temprana puede tardar años en demostrar beneficio real, seguridad a largo plazo y acceso razonable. Yo sería especialmente cauto con cualquier promesa que hable de “frenar” la enfermedad sin mostrar datos robustos y reproducibles. En enfermedades raras, el entusiasmo sin evidencia puede hacer tanto ruido como esperanza.
En cuanto al pronóstico, la evolución es progresiva y depende mucho del subtipo. Muchas personas afectadas llegan a la adolescencia y algunas a la adultez temprana, aunque los casos más severos empeoran antes; el tipo A suele ser el más precoz y agresivo. Con el avance de la enfermedad pueden aparecer pérdida de movilidad, convulsiones, trastornos del sueño cada vez más marcados y dificultades para tragar. Ese es el motivo por el que una detección temprana no solo importa para el diagnóstico, sino para planificar con tiempo todo lo demás.
Si yo tuviera que resumir la parte bioética en una frase, sería esta: cuanto más rara y progresiva es la enfermedad, más obligación tenemos de explicar bien qué se sabe, qué no se sabe y qué opciones reales existen. Esa claridad no quita esperanza; la vuelve útil. Y es la mejor manera de llegar al final con una idea práctica y no solo con un nombre médico.
Lo que yo vigilaría si tuviera que resumirlo para una familia
Si una familia me pidiera una guía breve, les diría que no pierdan de vista tres cosas: regresión del desarrollo, alteraciones persistentes del sueño y cambios de conducta que no encajan con una explicación simple. Cuando esos signos se combinan con infecciones repetidas, diarrea, torpeza motora o pérdida auditiva, merece la pena acelerar la derivación a genética clínica o neurología pediátrica.
- No normalizar la pérdida de palabras o habilidades ya adquiridas.
- No separar “conducta” de “neurología” si hay varios síntomas juntos.
- Pedir consejo genético si hay un caso familiar o si se confirma un subtipo.
- Plantear apoyos escolares y sociales desde temprano, no cuando el agotamiento ya es total.
La clave no es perseguir un diagnóstico por ansiedad, sino evitar meses o años perdidos. Cuanto antes se identifique la enfermedad, antes se puede ordenar el seguimiento, anticipar complicaciones y tomar decisiones reproductivas con información real. En una patología como esta, llegar pronto no lo resuelve todo, pero cambia mucho más de lo que parece.