Las trisomías más comunes en humanos no forman un bloque uniforme: unas son compatibles con la vida y otras suelen interrumpir el embarazo muy pronto. Si uno entiende bien el origen del error cromosómico, el tipo de trisomía y la prueba que la detecta, la interpretación clínica cambia por completo. En genética, ese matiz es la diferencia entre un hallazgo que solo orienta y un diagnóstico que realmente modifica el seguimiento.
Lo esencial para entender las trisomías más frecuentes
- Una trisomía significa que hay tres copias de un cromosoma en lugar de dos.
- Las más relevantes en nacidos vivos son trisomía 21, 18 y 13; entre los cromosomas sexuales destacan 47,XXY, 47,XXX y 47,XYY.
- La causa más habitual es la no disyunción, un fallo en la separación cromosómica durante la meiosis.
- El cribado prenatal estima riesgo; el diagnóstico se confirma con estudios como la biopsia corial, la amniocentesis o el cariotipo.
- La gravedad depende mucho de si la trisomía es completa, en mosaico o por translocación.
Cómo aparece una trisomía durante la división celular
La forma más útil de entender este tema es empezar por la división celular. En una célula humana normal hay 46 cromosomas, organizados en 23 pares. Si durante la formación de óvulos o espermatozoides algo falla en la separación de esos cromosomas, puede generarse un gameto con una copia de más y, tras la fecundación, un embrión con 47 cromosomas en lugar de 46.
Ese fallo se llama no disyunción, y consiste en que los cromosomas no se separan donde deben. Si ocurre en la meiosis I, no se separan los cromosomas homólogos; si ocurre en la meiosis II, el problema está en las cromátidas hermanas. El resultado práctico es parecido: un gameto anómalo que, al unirse con otro normal, produce una trisomía.
La no disyunción no ocurre siempre en el mismo momento
Cuando el error aparece antes de la fecundación, la trisomía suele estar presente en todas las células del individuo. Si el fallo se produce después, en una división embrionaria temprana, aparece mosaicismo: unas células tienen la trisomía y otras no. Ese detalle importa mucho porque el mosaico suele dar cuadros más variables y, en algunos casos, más leves.
También existen translocaciones, en las que el material extra no aparece como un cromosoma independiente, sino pegado a otro cromosoma. Eso cambia el consejo genético y, sobre todo, el riesgo de repetición familiar. Yo no interpretaría nunca una trisomía sin preguntar si es libre, en mosaico o por translocación.
Lee también: Cromosoma X - Clave en genética y salud. ¡Entiende su rol!
El mosaico y la translocación cambian mucho la lectura clínica
La idea clave aquí es simple: el nombre de la trisomía no lo dice todo. Dos personas con la misma etiqueta citogenética pueden tener evoluciones muy distintas si una presenta un mosaico parcial y la otra una trisomía completa. Por eso, en genética clínica, el contexto pesa casi tanto como el número del cromosoma implicado.
Con esta base ya se entiende por qué unas trisomías son relativamente compatibles con la vida y otras no. El siguiente paso es ordenar cuáles son las que realmente vemos con más frecuencia en nacidos vivos.
Cuáles son las trisomías más frecuentes en humanos
Si yo tuviera que ordenar este tema con una regla práctica, separaría primero las trisomías autosómicas viables y después las aneuploidías de los cromosomas sexuales. No tienen el mismo peso clínico ni la misma frecuencia, y conviene no mezclar ambas cosas como si fueran equivalentes.
| Condición | Cromosoma extra | Frecuencia aproximada | Lectura clínica rápida |
|---|---|---|---|
| Trisomía 21 o síndrome de Down | 21 | 1 de cada 700 recién nacidos | Es la trisomía autosómica viable más frecuente. La forma libre representa la mayoría de los casos; existen también formas por translocación y mosaico. |
| Trisomía 18 o síndrome de Edwards | 18 | 1 de cada 5.000 nacidos vivos | Es mucho más grave. Muchas gestaciones no llegan a término y, en los nacidos vivos, el pronóstico suele ser muy limitado. |
| Trisomía 13 o síndrome de Patau | 13 | 1 de cada 16.000 recién nacidos | También es una trisomía grave, con afectación multiorgánica importante y supervivencia reducida. |
| 47,XXY o síndrome de Klinefelter | X | 1 de cada 650 varones recién nacidos | Es la aneuploidía sexual más frecuente. Suele pasar desapercibida hasta la pubertad o la edad adulta. |
| 47,XXX o trisomía X | X | 1 de cada 1.000 recién nacidas | Con frecuencia da pocos rasgos físicos llamativos y se diagnostica tarde o nunca. |
| 47,XYY | Y | 1 de cada 1.000 recién nacidos | Suele tener una expresión muy variable y también está infradiagnosticada. |
Hay una matización importante: si hablamos con rigor, XXY, XXX y XYY no son trisomías autosómicas, sino aneuploidías de los cromosomas sexuales. Aun así, en las búsquedas generales suelen entrar en el mismo saco porque comparten la idea de “un cromosoma de más”.
Y hay otro matiz que cambia la perspectiva: cuando ampliamos la mirada a las pérdidas gestacionales, aparecen trisomías como la 16, muy frecuentes en abortos espontáneos y prácticamente incompatibles con un embarazo a término. Eso explica por qué la lista de trisomías que vemos en nacidos vivos es corta: muchas no llegan tan lejos.
Con el mapa ya ordenado, la pregunta lógica es por qué unas alteraciones dan cuadros severos y otras pasan casi inadvertidas.
Qué diferencias clínicas importan de verdad
La etiqueta citogenética no basta para prever el impacto clínico. Yo suelo fijarme en cuatro variables: el cromosoma afectado, si la trisomía es completa o en mosaico, si existe translocación y si hablamos de un autosoma o de un cromosoma sexual.
- En las trisomías autosómicas, el exceso de material genético altera muchos órganos a la vez. Por eso la trisomía 18 y la 13 suelen ser mucho más graves que la 21.
- En las trisomías sexuales, el cuadro suele ser más variable y a menudo más leve. Aquí ayuda la inactivación del cromosoma X, un mecanismo por el que parte del exceso de X queda silenciado en las células femeninas.
- En el mosaico, no todas las células tienen la misma dotación cromosómica. Eso puede suavizar el fenotipo, pero también hace más difícil anticipar la evolución solo con el nombre del diagnóstico.
- En las formas por translocación, el material extra está reorganizado. Esto no solo cambia la clínica; también puede cambiar el riesgo de recurrencia en futuros embarazos.
Ese contraste es importante porque evita dos errores frecuentes: asumir que todas las trisomías son igual de graves y asumir, por el contrario, que una trisomía sexual “no cuenta” por ser más leve. Ninguna de las dos ideas es correcta.
Con esas diferencias en mente, ya tiene sentido pasar a lo que más preocupa a muchas familias: cómo se detectan estas alteraciones y qué valor real tiene cada prueba.
Cómo se detectan en el embarazo y después del nacimiento
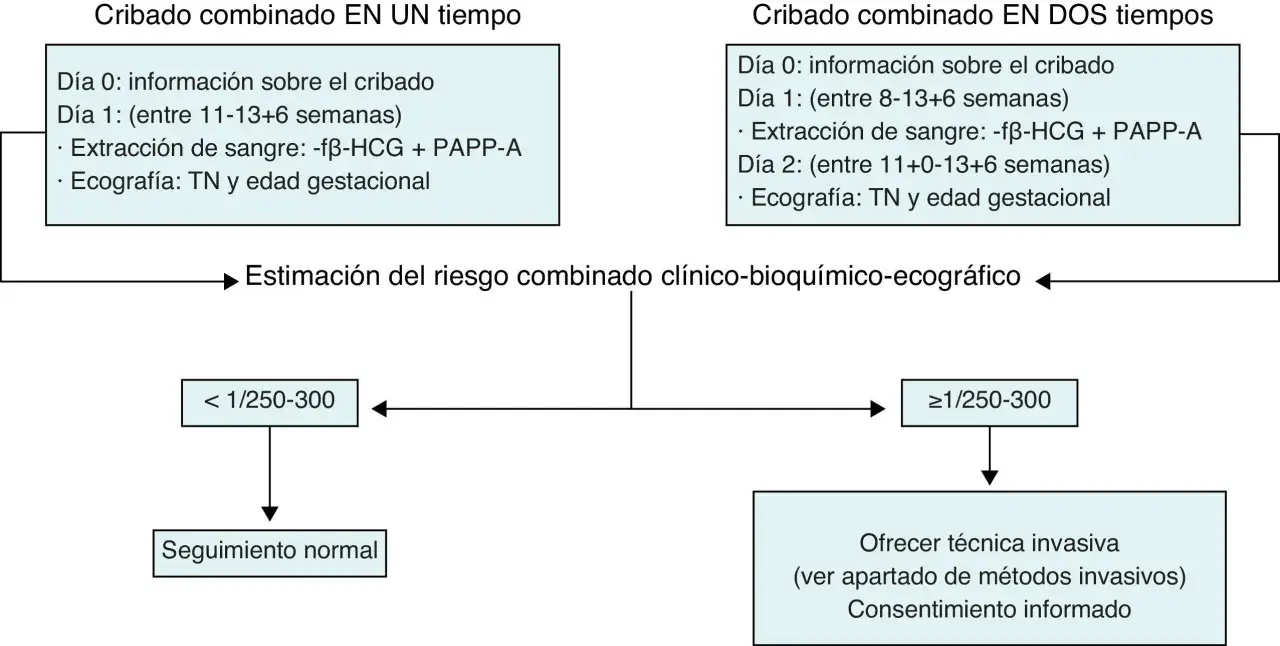
Yo insisto mucho en una distinción que evita confusiones: cribado no es lo mismo que diagnóstico. El cribado estima probabilidad; el diagnóstico confirma. Y en trisomías, esa diferencia no es un detalle técnico, sino el punto que separa una sospecha de una certeza clínica.
| Prueba | Qué aporta | Cuándo suele hacerse | Qué hay que tener claro |
|---|---|---|---|
| ADN fetal libre en sangre materna o cfDNA/NIPT | Estima el riesgo de trisomía 21, 18 y 13, y en algunos paneles también de aneuploidías sexuales | Desde la semana 10 | Es un cribado muy útil, pero no confirma por sí solo el diagnóstico. |
| Cribado combinado del primer trimestre | Une ecografía y analítica para calcular riesgo | Entre las semanas 10 y 13 | Sirve para orientar, no para cerrar el diagnóstico. |
| Biopsia corial (CVS) | Estudio directo de las vellosidades coriales, que comparten el material cromosómico del feto | Entre las semanas 10 y 13 | Es diagnóstica, pero es una prueba invasiva. |
| Amniocentesis | Analiza células del líquido amniótico | Entre las semanas 15 y 20 | También es diagnóstica y se usa para confirmar o descartar una alteración cromosómica. |
| Cariotipo posnatal | Confirma el número y la estructura de los cromosomas | Tras el nacimiento o cuando hay sospecha clínica | Es la referencia clásica para ver una trisomía y distinguir entre formas libres, mosaico o translocación. |
La práctica clínica actual ha afinado mucho el cribado prenatal, pero la lógica sigue siendo la misma: primero se estima riesgo y, si hace falta, después se confirma. Si el resultado sale alterado, el siguiente paso no debería ser improvisar, sino revisar qué prueba se hizo, qué cromosoma está implicado y si hace falta una confirmación invasiva.
En la consulta, además, yo no me quedaría solo en “positivo” o “negativo”. Preguntaría por el contexto ecográfico, por la edad gestacional y por la posibilidad de mosaico o translocación. Esa información cambia el pronóstico y también el consejo genético.
Una vez que la alteración queda confirmada, la cuestión deja de ser solo diagnóstica y pasa a ser de seguimiento, pronóstico y toma de decisiones informadas.
Qué cambia en el seguimiento y el consejo genético
Cuando una trisomía se confirma, no existe una corrección de fondo del cromosoma sobrante. El manejo es clínico y multidisciplinar: seguimiento pediátrico, cardiológico, endocrino, del desarrollo, del lenguaje y, cuando procede, apoyo a la fertilidad o a la adolescencia. La meta no es “arreglar” el cariotipo, sino anticipar complicaciones y acompañar bien.
- En trisomía 21, el seguimiento suele centrarse en corazón, tiroides, audición, visión, desarrollo motor y del lenguaje.
- En trisomía 18 y 13, el pronóstico depende mucho de las malformaciones asociadas y de la posibilidad de cuidados intensivos y apoyo paliativo o neonatal, según el caso y las decisiones familiares.
- En XXY, el consejo suele abordar hipogonadismo, fertilidad, desarrollo puberal y dificultades de aprendizaje o lenguaje.
- En XXX y XYY, la variabilidad es alta y muchas personas llevan una vida funcional sin diagnóstico hasta etapas tardías.
También importa el riesgo de repetición. Si la trisomía fue un evento aislado de no disyunción, el riesgo puede ser bajo, aunque la edad materna siga influyendo. Pero si existe una translocación equilibrada en uno de los progenitores, la situación cambia y el estudio familiar puede ser necesario. Aquí el consejo genético no es decorativo: evita falsas seguridades y también alarmas injustificadas.
Hay un punto bioético que no conviene maquillar. Cuando la alteración se detecta en embarazo, la familia no solo recibe un dato biológico; recibe también una información que puede condicionar decisiones complejas y muy personales. En esos momentos, informar con precisión y sin dramatismo es tan importante como saber leer el informe.
Si reviso un caso real, nunca me quedo en la etiqueta del síndrome. Miro si es completa, en mosaico o por translocación; verifico qué prueba lo demostró; y después traduzco eso a un pronóstico razonable, no a una predicción absoluta.
La clave práctica no es solo el cromosoma, sino el contexto del hallazgo
Si tengo que dejar una idea útil, es esta: una trisomía no se interpreta bien por el nombre solo, sino por el conjunto formado por cromosoma implicado, tipo de alteración y prueba diagnóstica. Ese trío explica por qué algunas alteraciones son relativamente frecuentes y compatibles con la vida, mientras otras aparecen sobre todo en gestaciones que no llegan a término.
También conviene recordar que las trisomías sexuales suelen estar más infradiagnosticadas que las autosómicas graves. No porque sean “menos reales”, sino porque sus signos pueden ser discretos y mezclarse con variaciones normales del desarrollo. En genética, lo silencioso no siempre es benigno; a veces solo está peor reconocido.
Cuando alguien me pide una visión práctica del tema, yo la reduzco a dos pasos: primero, distinguir si hablamos de cribado o de diagnóstico; después, leer el resultado en su contexto clínico y familiar. Con eso ya se evita la mayoría de interpretaciones erróneas y se gana una base sólida para decidir el siguiente paso con calma.