Hablar del genoma humano completo es hablar del mapa base de nuestra biología: la secuencia que explica por qué somos parecidos, por qué también somos diferentes y hasta dónde puede llegar hoy la genética clínica. En este artículo explico qué contiene realmente esa información, por qué costó tanto cerrarla y qué cambia cuando pasa de ser un borrador de referencia a una herramienta útil para la medicina personalizada.
También verás qué diferencia hay entre un exoma, una secuencia íntegra y un pangenoma, qué tipo de variantes detecta mejor cada enfoque y dónde empiezan los límites interpretativos. Si el tema te interesa por salud, investigación o bioética, aquí tienes una visión clara y práctica.
Lo esencial en pocas líneas
- Una copia del genoma humano contiene unos 3 mil millones de bases repartidas en 23 pares de cromosomas.
- Solo alrededor del 1,5% codifica proteínas; el resto regula, organiza o repite secuencias con funciones distintas.
- La parte difícil no era leer el ADN, sino ensamblarlo en regiones repetitivas como telómeros y centrómeros.
- La referencia humana ha pasado de un borrador lineal a una secuencia sin huecos y, después, a un pangenoma más diverso.
- En clínica, el valor real está en variantes raras, estructurales y farmacogenéticas, no solo en genes clásicos.
- Una secuencia no sustituye la historia clínica, el fenotipo, la epigenética ni el consejo genético.
Qué significa una secuencia de referencia realmente completa
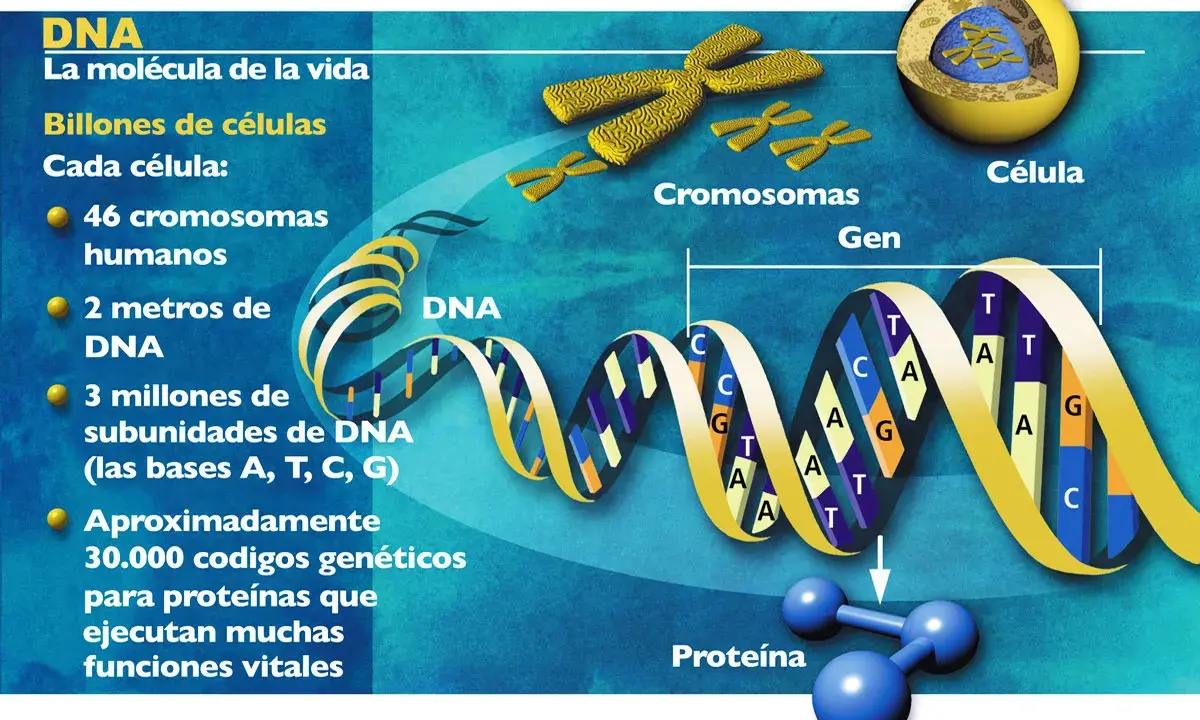
Si yo tuviera que explicarlo sin tecnicismos, diría que se trata de la secuencia de ADN de referencia que reúne todas las instrucciones heredables de nuestra especie. El dato clave no es solo el tamaño: una copia del genoma humano contiene unos 3 mil millones de bases y dos personas comparten más del 99% de esa secuencia.
Cuando hablo de un genoma humano completo, me refiero a una secuencia capaz de cubrir todo eso sin huecos importantes: genes, regiones reguladoras, intrones, telómeros, centrómeros y ADN repetitivo. Los genes codificadores de proteínas son solo alrededor de 20.000, y la parte que realmente codifica proteínas ocupa cerca del 1,5% del total; el resto no es “basura”, sino un territorio con funciones de estructura, regulación y organización muy distintas.
Aquí conviene hacer una distinción que suele pasarse por alto: genoma no es lo mismo que genes. Los genes son unidades funcionales; el genoma es el conjunto completo que los contiene y los regula. Esa diferencia importa mucho, porque una alteración fuera de un gen también puede cambiar la biología de una persona. Y justo ahí empieza el problema técnico que durante años mantuvo la secuencia incompleta.
Por qué fue tan difícil cerrarlo
La dificultad no era solo leer ADN, sino reconstruirlo en el orden correcto. Imagina un libro de miles de millones de letras del que solo puedes fotografiar fragmentos: si además muchas páginas se repiten casi igual, el ensamblaje se vuelve un rompecabezas de altísima precisión. Durante mucho tiempo, la secuenciación por lecturas cortas permitió avanzar rápido, pero dejaba zonas enteras sin resolver o mal colocadas.
- Las repeticiones confundían al ensamblador: no era obvio qué fragmento pertenecía a qué región.
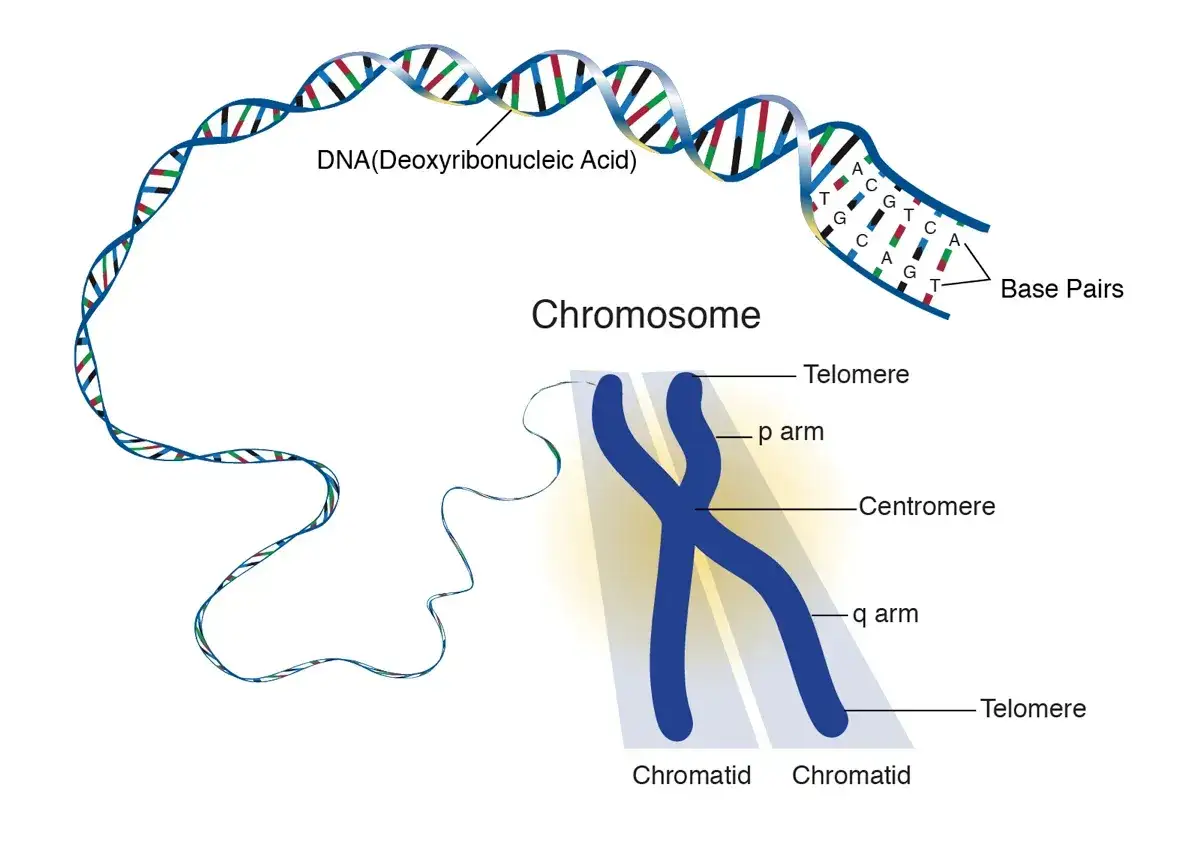
- Los telómeros, que son los extremos protectores de los cromosomas, y los centrómeros, que ayudan a separarlos durante la división celular, eran especialmente difíciles de leer.
- El último tramo resultó desproporcionadamente duro: cerrar ese 8% final llevó casi tanto esfuerzo como completar el 92% anterior.
La salida llegó con lecturas largas, capaces de atravesar regiones repetitivas completas y de anclar mejor las piezas del puzle. Ese cambio técnico no solo llenó huecos: también reveló variantes y secuencias que antes estaban ocultas, justo las que más interés tienen para la biología molecular y el diagnóstico. Una vez superado ese obstáculo, la pregunta dejó de ser cómo leer el ADN y pasó a ser qué referencia usar para interpretarlo bien.
Genoma, exoma y pangenoma no sirven para lo mismo
| Enfoque | Qué cubre | Para qué suele ser mejor | Limitación principal |
|---|---|---|---|
| Exoma | Solo las regiones codificantes, cerca del 1-2% del ADN | Variantes en genes que producen proteínas | Deja fuera muchas regiones reguladoras y gran parte de las variantes estructurales |
| Secuencia completa | Prácticamente toda la secuencia nuclear | Casos complejos, regiones no codificantes, variantes grandes o repetitivas | Genera más datos y exige una interpretación más difícil |
| Pangenoma | Una referencia construida con varios genomas, no con uno solo | Reducir el sesgo de una referencia única y representar mejor la diversidad | No sustituye por sí mismo al análisis clínico ni está igual de integrado en todos los flujos de trabajo |
Esta comparación es importante porque mucha gente mezcla conceptos que no son equivalentes. Un exoma puede bastar cuando la sospecha clínica apunta claramente a genes codificadores; una secuencia completa gana terreno cuando el caso es oscuro, cuando se sospechan variantes estructurales o cuando un resultado parcial no explica el cuadro. Yo suelo verlo así: cuanto más difícil es el caso, menos sentido tiene quedarse solo con una foto recortada.
El pangenoma, por su parte, no es una prueba que se pida al paciente, sino una forma más inteligente de construir la referencia. En vez de usar una sola secuencia “promedio”, incorpora diversidad de diferentes personas y reduce el sesgo de referencia, que es el problema de intentar entender a toda la especie con un modelo demasiado estrecho. La primera versión pública reunió secuencias de 47 personas, equivalentes a 94 genomas haploides, y añadió más de 100 millones de bases frente a la referencia anterior. Ese salto no busca competir con la secuencia completa como prueba clínica; busca que la referencia con la que comparo los datos sea menos sesgada y más representativa.
Qué cambia en medicina personalizada y diagnóstico
La secuencia completa no vale por sí sola como diagnóstico, pero sí puede cambiar mucho la forma en que busco una explicación clínica. Su mayor valor aparece cuando necesito salir de un caso que no encaja en paneles limitados o cuando quiero ver más allá de los cambios en proteínas.
- Enfermedades raras sin diagnóstico: una secuencia completa puede detectar variantes intrónicas, reguladoras o estructurales que un exoma no ve con facilidad.
- Farmacogenética: ayuda a entender por qué una persona metaboliza peor o mejor ciertos fármacos, siempre que haya evidencia clínica suficiente para actuar sobre ese hallazgo.
- Oncología: en cáncer, la secuencia tumoral y la heredada no son lo mismo; distinguirlas evita confundir una predisposición germinal con una mutación adquirida por el tumor.
- Consejo genético familiar: cuando hay un hallazgo relevante, permite valorar riesgos en otros familiares con más precisión, aunque nunca de forma automática.
También hay una diferencia económica y práctica que conviene no maquillar. Leer más datos ya no es el gran cuello de botella; interpretarlos bien lo es. La secuenciación se ha abaratado mucho, pero el tiempo y el coste de análisis, validación y comunicación clínica siguen pesando bastante más que el simple acto de generar archivos de ADN.
En la práctica, el mejor resultado aparece cuando la secuencia se cruza con fenotipo, historia familiar y pruebas complementarias. Si una variante sale en aislamiento, sin contexto clínico, la utilidad baja enseguida. Por eso la medicina personalizada de verdad no consiste en tener más letras, sino en saber qué letras cambian una decisión.
Dónde se suelen malinterpretar los resultados
Este es el punto que más me interesa desde la bioética: una secuencia no es una sentencia. Encontrar una variante no significa que una enfermedad vaya a aparecer, ni que su gravedad sea inevitable, ni que dos personas con la misma alteración vayan a evolucionar igual. La biología humana es probabilística, no mecánica.
- Una variante no equivale a un destino: hay penetrancia incompleta, es decir, no todas las personas con la misma variante desarrollan el mismo rasgo, y además influyen modificadores genéticos y entorno.
- Un hallazgo incierto no debe sobreinterpretarse: muchas variantes siguen clasificadas como de significado incierto.
- La representación poblacional importa: si una referencia está sesgada hacia ciertos orígenes, interpreta peor a otras poblaciones.
- La secuencia no sustituye la biología funcional: epigenética, expresión génica, proteínas y ambiente siguen contando.
- La privacidad importa: los datos genéticos son sensibles y, bien gestionados, deben ir acompañados de consentimiento claro y políticas de uso responsables.
- Los hallazgos incidentales existen: a veces aparece información relevante que no motivó la prueba y exige decidir qué se comunica y cómo.
También conviene separar la promesa científica del entusiasmo comercial. Decir que se puede secuenciar el ADN no es lo mismo que afirmar que se puede explicar toda la salud de una persona. El genoma ayuda mucho, pero no lo explica todo. Cuando esa frontera no se respeta, aparecen falsas expectativas y decisiones mal apoyadas.
Lo que conviene recordar antes de convertir ADN en decisiones
Si tuviera que dejar tres ideas finales, me quedaría con estas: el genoma aporta mapa, no destino; la referencia humana sigue perfeccionándose para representar mejor la diversidad; y la utilidad clínica real aparece cuando la secuencia se interpreta con contexto, no como un resultado aislado.
- Una secuencia completa amplía lo que puedes ver, pero no resuelve por sí sola todas las preguntas clínicas.
- El valor aumenta cuando el caso es complejo, hereditario, poco claro o sospechoso de variantes estructurales.
- La interpretación mejora si se integra con asesoramiento genético, datos familiares y confirmación funcional cuando haga falta.
- La bioética no es un añadido decorativo: consentimiento, privacidad y equidad cambian el modo en que la genómica debe aplicarse.