Lo fundamental sobre su origen genético
- La causa directa es una expansión de repeticiones CAG en el gen HTT, situado en el cromosoma 4.
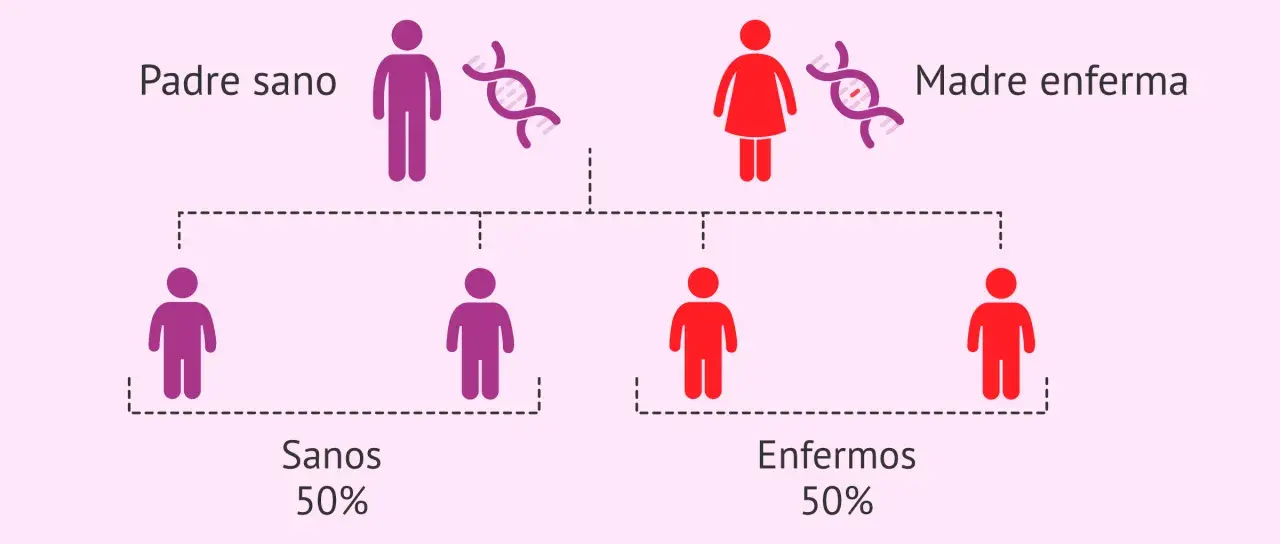
- La herencia es autosómica dominante: si uno de los progenitores tiene la expansión patógena, cada hijo tiene un 50% de probabilidad de heredarlo.
- No es una enfermedad causada por hábitos, estrés, alimentación o ejercicio; el origen es genético.
- La cantidad de repeticiones influye en el riesgo y en la edad de inicio, pero no permite predecir el calendario exacto de síntomas.
- El daño cerebral afecta sobre todo a circuitos del estriado y la corteza, por eso aparecen síntomas motores, cognitivos y psiquiátricos.
- El consejo genético es clave para interpretar resultados, valorar pruebas predictivas y tomar decisiones reproductivas con información real.
La causa real está en un cambio del gen HTT
Yo suelo resumir el origen de esta enfermedad en una idea muy concreta: una expansión anómala de repeticiones CAG en el gen HTT altera la proteína huntingtina y la convierte en tóxica para ciertas neuronas. El gen HTT contiene una secuencia que, en condiciones normales, tiene un número limitado de repeticiones; cuando ese tramo se alarga demasiado, la proteína resultante cambia de comportamiento y empieza a dañar el sistema nervioso de forma progresiva.
La forma más útil de entenderlo es separar tres niveles: el gen, la proteína y el cerebro. El problema arranca en el ADN, continúa en la proteína huntingtina mutada y termina expresándose en el deterioro de circuitos neuronales que regulan el movimiento, la memoria, la conducta y el control ejecutivo.
| Repeticiones CAG en HTT | Interpretación habitual | Qué significa en la práctica |
|---|---|---|
| 26 o menos | Rango normal | No se considera causante de Huntington |
| 27 a 35 | Alelo intermedio | La persona suele no desarrollar la enfermedad, pero el tramo puede expandirse en la descendencia |
| 36 a 39 | Penetrancia reducida | Puede haber enfermedad o no; el riesgo es variable |
| 40 o más | Penetrancia completa | Riesgo muy alto de desarrollar la enfermedad a lo largo de la vida |
Esta tabla ayuda a ordenar la parte genética, pero no conviene leerla como una sentencia exacta. El número de repeticiones orienta, no dicta con precisión cuándo aparecerán los síntomas ni cómo evolucionará cada persona. Esa parte depende de otros moduladores biológicos que veremos enseguida.
Qué le hace al cerebro la huntingtina alterada
La huntingtina mutada no daña una sola vía; altera varias a la vez. A nivel celular, hablamos de una toxicidad de ganancia de función: la proteína no solo deja de comportarse con normalidad, sino que además adquiere efectos perjudiciales. Esa combinación explica por qué la enfermedad es tan compleja y por qué no basta con pensar en un único mecanismo.
Las neuronas más vulnerables son las del estriado, una zona muy implicada en el control del movimiento, y también se ven afectadas redes corticales relacionadas con el pensamiento y la conducta. Por eso la clínica no se limita a los temblores o a los movimientos involuntarios: también aparecen cambios de personalidad, dificultad para planificar, lentitud mental y síntomas depresivos o irritabilidad.
- Transcripción génica alterada: la célula fabrica proteínas en proporciones menos equilibradas, y eso reduce su capacidad de resistencia al daño.
- Disfunción mitocondrial: la producción de energía se vuelve menos eficiente, algo crítico en neuronas que consumen mucha energía.
- Autofagia defectuosa: los sistemas de limpieza celular eliminan peor los desechos, y se acumulan proteínas anómalas.
- Alteración del tráfico axonal: el transporte interno de materiales entre el cuerpo neuronal y sus prolongaciones se vuelve menos preciso.
- Desregulación sináptica y del calcio: la comunicación entre neuronas se vuelve más inestable y el circuito pierde precisión.
En la práctica, todo eso se traduce en degeneración progresiva. No es un daño brusco ni uniforme, sino un desgaste que tarda años en hacerse visible. Y precisamente por eso la siguiente pregunta importante no es solo qué causa la enfermedad, sino por qué en unas familias aparece antes y en otras mucho más tarde.
Cómo se hereda y qué riesgo real tiene la familia
La enfermedad de Huntington sigue un patrón autosómico dominante. Eso significa que basta una sola copia alterada del gen para que la persona tenga riesgo de desarrollar la enfermedad. Si un progenitor porta una expansión patogénica, cada hijo tiene un 50% de probabilidad de heredarla.
Una idea que intento corregir siempre es la culpa: nadie “elige” transmitir este cambio genético. La herencia no depende de lo que la madre o el padre hayan hecho durante el embarazo, ni de su alimentación, ni de su nivel de estrés. El factor determinante es la presencia o ausencia de la expansión en el material genético.
Hay, además, un matiz importante: las repeticiones CAG pueden comportarse de forma inestable cuando pasan de una generación a otra. Eso explica que algunas familias vean expansiones más largas en la descendencia, sobre todo cuando la transmisión es paterna. No es una regla absoluta, pero sí un fenómeno bien conocido en genética clínica.
- Si el alelo es intermedio, la persona puede no tener síntomas, pero sí transmitir un tramo más largo a sus hijos.
- Si el alelo está en el rango de penetrancia reducida, el futuro clínico es menos predecible.
- Si la expansión es claramente patogénica, el riesgo de desarrollar la enfermedad es muy alto.
Esta parte es clave para entender por qué el consejo genético no se limita a “decir si sí o si no”. También ayuda a interpretar riesgos familiares, valorar opciones reproductivas y evitar lecturas simplistas que luego generan más ansiedad que información útil.
Por qué la edad de inicio cambia tanto
Uno de los errores más frecuentes es pensar que el número de repeticiones CAG determina exactamente cuándo empezarán los síntomas. No es así. Sí existe una relación general: cuantas más repeticiones, mayor tendencia a un inicio más temprano. Pero entre una persona y otra siguen influyendo otros factores, genéticos y biológicos, que hacen imposible dar una fecha precisa.
En la práctica, los síntomas suelen aparecer en la edad adulta, con frecuencia entre los 30 y los 50 años. Existe también una forma juvenil, que comienza antes y suele asociarse con expansiones más grandes. En esos casos, el curso clínico puede ser distinto y a veces más agresivo, aunque tampoco hay un umbral único que lo explique todo.
La anticipación genética es el fenómeno por el cual la enfermedad puede debutar antes en una generación que en la anterior, sobre todo cuando la expansión se agranda al transmitirse. Aquí conviene ser preciso: no significa que la enfermedad “empeore porque sí”, sino que el tramo repetitivo puede ser más inestable durante la transmisión familiar.
Yo lo explico de forma simple: la genética fija la dirección del riesgo, pero no marca con exactitud el ritmo. Esa diferencia es importante porque evita falsas certezas y ayuda a leer mejor los resultados de una prueba molecular.
Lo que no la causa y por qué conviene decirlo en voz alta
Cuando alguien oye hablar de Huntington, a menudo busca una explicación externa que suene más familiar: el estrés, una caída, una infección, la dieta, una etapa emocional difícil. Ninguna de esas cosas causa la enfermedad. Pueden influir en cómo se siente la persona o en cómo afronta los síntomas, pero no generan la mutación que la produce.
Tampoco estamos ante una enfermedad contagiosa, ni autoinmune, ni provocada por el estilo de vida. Esa precisión importa más de lo que parece, porque reduce culpa, evita interpretaciones erróneas y centra la conversación en lo que sí tiene peso clínico: la genética, el seguimiento neurológico y el apoyo familiar.
- No la causa el estrés emocional.
- No la causa una mala alimentación.
- No la causa el ejercicio físico.
- No la causa una infección habitual.
- No la causa un golpe aislado en la cabeza.
Lo que sí puede ocurrir es que una persona tenga la mutación durante años sin saberlo, y que los primeros síntomas se confundan con depresión, torpeza, cambios de humor o problemas de concentración. Esa ambigüedad es una de las razones por las que el diagnóstico no debe basarse solo en impresiones clínicas.
Cómo se confirma hoy la alteración genética
La confirmación se hace con prueba genética molecular, que cuenta el número de repeticiones CAG en el gen HTT. En un contexto clínico adecuado, esa prueba puede confirmar la causa cuando hay síntomas compatibles y también puede servir como prueba predictiva en adultos en riesgo, siempre con asesoramiento previo.En la práctica, el proceso serio no empieza por el laboratorio, sino por la conversación clínica. Antes de pedir una prueba predictiva, conviene entender qué puede aportar un resultado positivo, qué no puede decir y qué impacto emocional, familiar y reproductivo puede tener. La información genética es poderosa, pero también tiene consecuencias.
- Valoración clínica: síntomas, exploración neurológica y antecedentes familiares.
- Consejo genético: explicación del alcance de la prueba, sus límites y sus efectos posibles.
- Análisis molecular del HTT: recuento de repeticiones CAG.
- Interpretación del resultado: rango normal, intermedio, penetrancia reducida o expansión patogénica.
- Plan de seguimiento: vigilancia, apoyo psicosocial y, si se desea, opciones reproductivas.
Qué cambia para la familia cuando se conoce la mutación
Conocer la causa genética cambia mucho más que una etiqueta diagnóstica. Cambia la forma de planificar hijos, de comunicar riesgos entre hermanos, de preparar apoyos futuros y de interpretar síntomas iniciales que antes parecían difusos. En una enfermedad como esta, la información no solo describe el presente: también reorganiza decisiones de largo recorrido.Una cosa que veo con frecuencia es la tentación de convertir el resultado en destino. No me parece una lectura útil. Saber que existe una expansión CAG no obliga a vivir en alarma permanente; obliga, más bien, a tomar decisiones mejor informadas y a usar el seguimiento neurológico y el consejo genético con criterio.
- Planificación familiar: valorar reproducción asistida, diagnóstico preimplantacional o alternativas personales.
- Comunicación entre parientes: decidir qué se comparte, cuándo y con qué apoyo profesional.
- Preparación psicológica: anticipar el impacto emocional de un resultado positivo o incierto.
- Seguimiento clínico: organizar una vigilancia más ordenada si hay síntomas o riesgo confirmado.
En menores asintomáticos, la prueba predictiva suele tratarse con especial prudencia porque la información genética tiene efectos de por vida y conviene preservar la autonomía futura de la persona. Ese punto, que a veces se pasa por alto, es una buena muestra de por qué genética y bioética deben ir juntas y no por separado.
Lo esencial para entender su origen sin simplificarlo
La causa de la enfermedad de Huntington no es misteriosa, pero sí exige precisión: una expansión CAG en HTT desencadena una cascada de toxicidad neuronal que afecta sobre todo al estriado y a la corteza. A partir de ahí se entiende la herencia dominante, el 50% de riesgo por cada embarazo cuando uno de los progenitores porta la mutación y la gran variabilidad en la edad de inicio.
Si tengo que dejar una idea final, es esta: el resultado genético orienta el riesgo, pero no agota la historia clínica de una persona ni la de su familia. Para leer bien esta enfermedad hace falta combinar biología molecular, neurología y consejo genético, porque ahí es donde la explicación deja de ser teórica y empieza a servir de verdad.
Por eso, ante antecedentes familiares o síntomas compatibles, lo razonable no es improvisar conclusiones, sino buscar una valoración especializada que interprete la mutación, el contexto familiar y las implicaciones prácticas con la calma que este tema exige.