La hemofilia no es una sola entidad, sino un grupo de trastornos hereditarios en los que falta un factor concreto de coagulación. Los tipos de hemofilia se distinguen por el factor afectado, pero en la práctica también importa cuánto cae su actividad y si el problema sigue un patrón ligado al cromosoma X o autosómico. En este artículo voy a ordenar esa clasificación, explicar qué cambia entre las formas A, B y C, y aterrizar lo que de verdad necesitas mirar en el diagnóstico, el tratamiento y el consejo genético.
Las claves para distinguir sus formas y actuar a tiempo
- Hemofilia A = déficit de factor VIII; es la forma más frecuente.
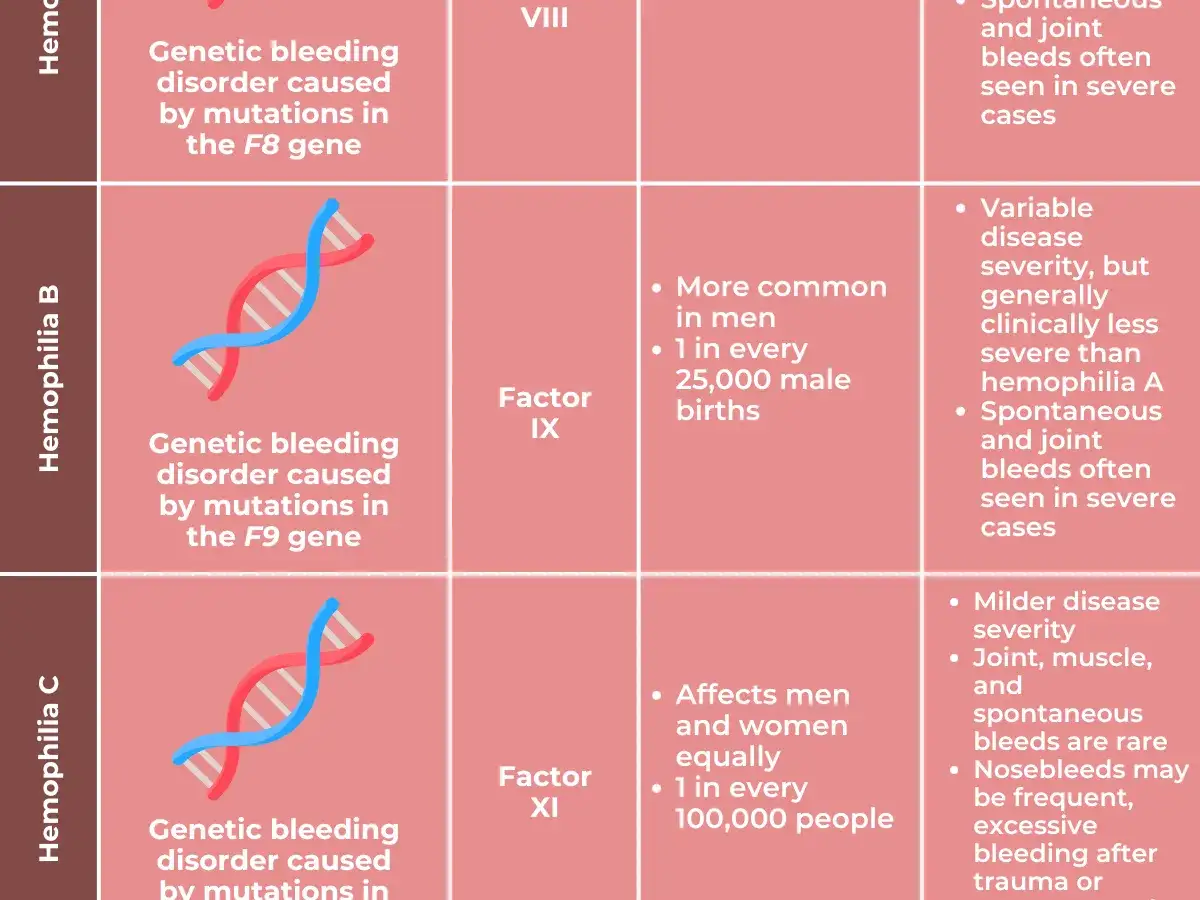
- Hemofilia B = déficit de factor IX; se parece mucho a la A en síntomas y manejo.
- Hemofilia C = déficit de factor XI; es más rara y suele dar un patrón de sangrado distinto.
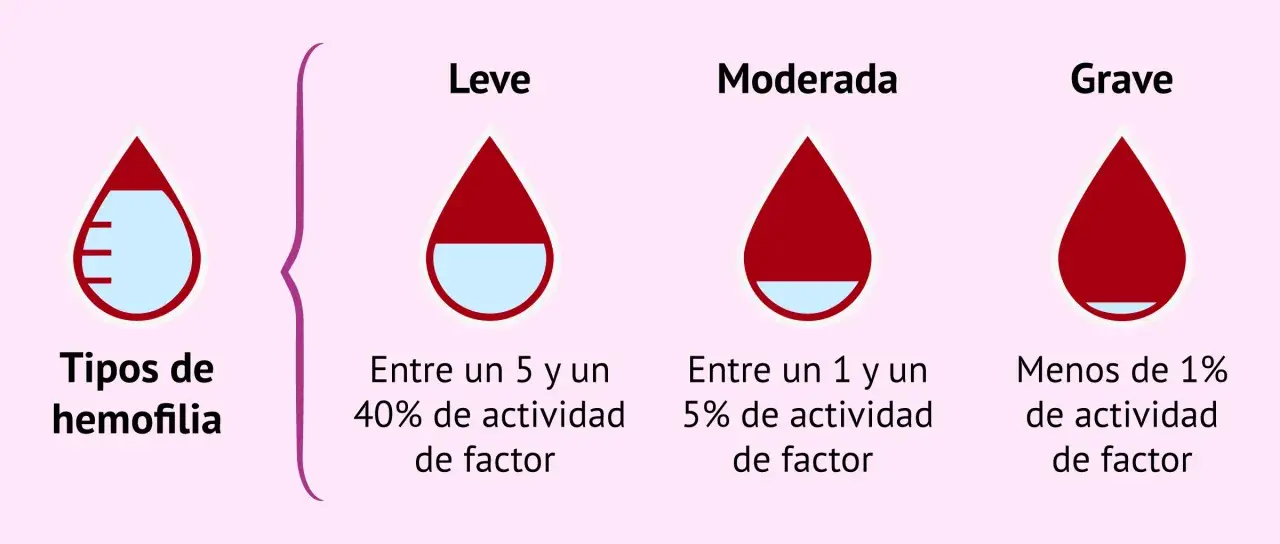
- La gravedad se clasifica por la actividad del factor: leve, moderada o grave.
- El diagnóstico real exige medir el factor concreto y, cuando conviene, estudiar la variante genética.
- La profilaxis y el consejo familiar cambian según el subtipo, la historia de sangrado y la edad.
Cómo se clasifica la hemofilia según el factor que falta
Yo suelo separar esta clasificación en dos planos: primero, el factor que falta; después, la intensidad del sangrado. Esa distinción evita un error muy común, que es asumir que todo lo que se llama hemofilia se comporta igual. En realidad, el nombre cambia según el factor deficiente, pero la conducta clínica depende también de cuánto factor queda funcional en sangre.
| Tipo | Factor deficiente | Patrón hereditario típico | Lo que suele destacar |
|---|---|---|---|
| Hemofilia A | Factor VIII | Ligada al cromosoma X | La forma más frecuente; sangrado articular y muscular en casos moderados o graves |
| Hemofilia B | Factor IX | Ligada al cromosoma X | Muy parecida a la A, pero causada por otra alteración genética |
| Hemofilia C | Factor XI | Generalmente autosómica recesiva | Más rara; suele notarse más tras cirugía, extracción dental o traumatismos |
Fuera de estas tres formas también existen otras deficiencias hereditarias de factores de coagulación, pero no todas se agrupan dentro de la hemofilia clásica. Esa precisión importa más de lo que parece, porque no solo cambia la etiqueta: cambia la herencia, el riesgo familiar y, en parte, el tipo de seguimiento que conviene planificar. Y precisamente ahí empieza la comparación útil entre A, B y C.
En qué se parecen y en qué se separan la A, la B y la C
La hemofilia A y la B se parecen tanto que, sin laboratorio, pueden confundirse con facilidad. Ambas suelen seguir una herencia ligada al cromosoma X, por eso los varones tienen más probabilidad de presentar síntomas relevantes y muchas mujeres aparecen como portadoras, aunque eso no significa que siempre estén asintomáticas. En la práctica, la diferencia real está en el gen y en el factor que se queda corto.
- Hemofilia A: déficit de factor VIII. Es la forma más frecuente y la que más a menudo se asocia al cuadro “clásico” de hemorragias articulares.
- Hemofilia B: déficit de factor IX. Clínicamente puede parecer una A, pero el defecto molecular es distinto y eso cuenta para el diagnóstico y el consejo genético.
- Hemofilia C: déficit de factor XI. Suele ser más variable, más rara y, en general, menos marcada por hemartrosis que las formas A y B.
La diferencia epidemiológica también ayuda a orientarse: la hemofilia A es varias veces más frecuente que la B, y en conjunto la hemofilia aparece en torno a 1 de cada 5.000 nacimientos de varones. Eso explica por qué muchas unidades tienen más experiencia acumulada con la A, aunque el esquema de estudio sea muy parecido en ambas.
La hemofilia C, en cambio, rompe un poco el patrón clásico. Al ser habitualmente autosómica recesiva, puede afectar a hombres y mujeres de forma similar, y además el sangrado no siempre guarda una relación lineal con el nivel del factor XI. Dicho de otro modo: el número ayuda, pero no siempre predice bien el comportamiento real del paciente. Esa idea nos lleva a la gravedad clínica, que es donde de verdad se decide cuánto riesgo hay.Cómo se mide la gravedad y qué síntomas orientan al tipo correcto
La severidad no se define por intuición, sino por la actividad residual del factor en sangre. Yo considero útil esa clasificación porque da un lenguaje común entre hematología, laboratorio y paciente, pero no debe leerse como una sentencia rígida: la historia de sangrado pesa mucho, sobre todo cuando hay hemofilia C o cuando el paciente ha aprendido a evitar situaciones de riesgo desde pequeño.
| Gravedad | Actividad del factor | Presentación típica |
|---|---|---|
| Leve | Más de 5% y menos de 40% | Sangrado prolongado tras cirugía, extracciones dentales o traumatismos; puede diagnosticarse tarde |
| Moderada | Entre 1% y 5% | Sangrado desproporcionado tras golpes menores; a veces aparecen sangrados espontáneos ocasionales |
| Grave | Menos de 1% | Hemorragias espontáneas, especialmente en articulaciones y músculos; inicio más temprano |
Los síntomas que más orientan son los hematomas fáciles, el sangrado prolongado tras una extracción dental, las epistaxis repetidas, los hematomas musculares y la hemartrosis, es decir, el sangrado dentro de la articulación. En mujeres, una señal que no conviene minimizar es la menstruación muy abundante o el sangrado posparto, porque a menudo se normaliza demasiado rápido y se tarda en conectar con un problema de coagulación.
Cuando yo veo un cuadro de sangrado repetido sin una explicación clara, no me quedo solo con el tipo de hemofilia: me pregunto si encaja mejor con una forma leve que ha pasado desapercibida o con una forma grave que necesita profilaxis desde ya. Esa diferencia es la que cambia el ritmo de seguimiento y prepara el terreno para el diagnóstico formal.
Cómo se confirma el diagnóstico sin perder tiempo ni pistas
No basta con pedir un análisis general y dar el caso por cerrado. El diagnóstico de verdad se construye por capas: primero, la historia clínica; después, las pruebas de coagulación; luego, la medición del factor concreto; y, si hace falta, el estudio genético y la búsqueda de inhibidores. Yo prefiero ese orden porque evita dos errores frecuentes: infravalorar un sangrado real y sobrediagnosticar a partir de un test aislado.
- Historia clínica y antecedentes familiares: sangrado tras cirugía, dentista, parto o pequeños traumatismos; también historia familiar de hemorragias.
- Pruebas de cribado: el TTPa suele orientar hacia un problema en la vía intrínseca de la coagulación; el TP suele mantenerse normal en las hemofilias A y B.
- Ensayo del factor específico: permite medir factor VIII, IX o XI y asignar el subtipo correcto.
- Estudio genético: confirma la variante, ayuda a identificar portadores y ordena el riesgo familiar.
- Estudio de inhibidores: se solicita si la respuesta al tratamiento es mala o si el paciente ha recibido factor de forma repetida.
La clave práctica es esta: un TTPa alterado orienta, pero no sentencia. Puede haber otras causas de sangrado o de alteración de la coagulación, y en un adulto sin antecedentes familiares también hay que pensar en procesos adquiridos. Por eso el laboratorio debe interpretarse con contexto clínico, no como una etiqueta aislada que resuelva todo por sí sola.
Qué tratamientos se usan hoy y cuándo cambia la estrategia
La base del tratamiento sigue siendo sencilla de enunciar y compleja de personalizar: recuperar la función del factor que falta, prevenir el daño articular y frenar los sangrados antes de que se compliquen. La idea de fondo no ha cambiado, pero sí ha cambiado mucho la forma de aplicarla, y eso se nota especialmente en pacientes jóvenes, en quienes hacen deporte y en quienes van a pasar por cirugía o procedimientos dentales.
- Reposición del factor VIII o IX: es la piedra angular en la hemofilia A y B, ya sea a demanda o en profilaxis.
- Desmopresina: puede servir en algunas formas leves de hemofilia A, pero no es una solución universal ni equivalente para la hemofilia B.
- Antifibrinolíticos: resultan útiles en sangrados de mucosas, sangrado dental y epistaxis, aunque no sustituyen al factor en una hemorragia mayor.
- Profilaxis: se usa cuando el objetivo es prevenir sangrados repetidos, sobre todo en formas graves o en pacientes con articulaciones ya dañadas.
- Emicizumab y otras estrategias no sustitutivas: han cambiado la prevención en hemofilia A, sobre todo cuando se busca un control más estable o cuando hay inhibidores.
La terapia génica ya forma parte de la conversación clínica en este campo, pero su encaje real sigue siendo selectivo y depende mucho del subtipo, del perfil del paciente y del centro que lo valora. Cuando el factor ya no explica todo, la genética termina de cerrar el mapa, y ahí es donde la siguiente pieza cobra sentido.
Qué aporta la genética al diagnóstico y al consejo familiar
Si hay una parte que me parece especialmente relevante en una web centrada en genética y medicina personalizada, es esta: la genética no solo nombra la enfermedad, también ordena el riesgo familiar. En las formas A y B, la herencia suele ser ligada al cromosoma X; eso explica por qué los varones presentan más a menudo el cuadro clínico típico y por qué algunas mujeres son portadoras sin síntomas, mientras que otras sí tienen sangrados porque su nivel de factor no es realmente normal. En la hemofilia C, la lógica cambia: al ser habitualmente autosómica recesiva, hombres y mujeres pueden verse afectados de forma parecida.
El estudio genético sirve para tres cosas muy concretas: confirmar la variante, detectar portadores y planificar embarazos con información realista. Eso puede incluir asesoramiento reproductivo, diagnóstico prenatal o estudio de familiares cuando existe una razón clínica clara. Yo aquí soy bastante directo: no se trata de “etiquetar” a una familia, sino de evitar sorpresas en cirugías, partos o extracciones dentales, donde una hemorragia mal preparada sí cambia el pronóstico.
También conviene no simplificar demasiado el papel de las mujeres. Algunas portadoras tienen menstruaciones abundantes, hematomas fáciles o sangrados prolongados tras procedimientos, aunque durante años se las haya dejado fuera del relato clásico de la hemofilia. Ese cambio de mirada importa, porque una portadora sintomática necesita seguimiento real, no una nota al margen.
Y hay una cuestión bioética de fondo: el valor de una prueba genética depende de que vaya acompañada de información clara, consentimiento y una conversación honesta sobre qué cambia y qué no cambia en la vida de la persona. Sin ese contexto, el resultado puede confundir más de lo que ayuda, especialmente cuando la familia está intentando tomar decisiones reproductivas o anticipar intervenciones médicas.
La lectura útil de un informe de coagulación
Si tuviera que dejar una regla práctica, sería esta: primero identifica el factor deficiente, después mide la gravedad y, por último, pregunta por la herencia. Ese orden evita errores frecuentes como mezclar hemofilia A con B, infravalorar una hemofilia C o asumir que un sangrado aislado descarta un problema genético.
La idea más útil no es memorizar letras, sino reconocer que la hemofilia cambia de forma según el factor implicado, el porcentaje de actividad residual y el contexto clínico. Si el sangrado es repetido, hay historia familiar o una cirugía está cerca, la valoración en una unidad especializada marca la diferencia entre reaccionar tarde y planificar con precisión.